Прионная болезнь это: Прионные болезни — причины, симптомы, диагностика и лечение
Прионы — страх и ужас будущего / Хабр
«Предрассветная дымка нехотя отступала по оврагам, проступали стебли ржи, переливающиеся под взмахами ветра. Птицы уже успели обрадоваться утру и ненавязчиво щебетали над ухом. Последние капли сна упали в чашку ароматного кофе. Приятно встречать диск солнца, растягивая заиндевевшие суставы и вглядываясь в даль. Кто это? Застыла мысль, когда взгляд скользнул на тропинку, бегущую из леса. Широкая улыбка озарила лицо. С первых движений он узнал ее. Только она могла двигаться с такой грацией и изяществом лани. Рука замерла на полпути к столу. Продолжая улыбаться, он вдруг резко повернулся и зашагал на кухню. Появились на столе еще одна чашка и поднос с ягодами. Аромат лавандового сиропа заполонил веранду. Хороший будет день, подумалось ему, приятный завтрак – уж точно.
Горсть малины быстро исчезала с подноса ягода за ягодой. Нежный женский голос рассказывал последние вести. За прошедшую неделю не поставили ни одного креста на городском кладбище. Мы дожили! — Выдох радости вырвался из уже скукоженных от возраста легких. Да!- ответила она ему. Эти чертовы четверть века. Четверть века, которые не оставили на твоем лице ни одного гладкого участка.
Вот такое начало фантастического рассказа прочитал я на досуге. В нем описывается новый вид биологического оружия. Ужасающий в своей разрушительной мощи. Людей охватывало оцепенение, когда они узнавали свою участь. Страх перед этой невидимой и неотвратимой напастью был хуже самой смерти.
«Почесуха»
Великобритания, первая половина 18 века. Туман рассеивается над зелеными сочными полями. Большое стадо овец не спеша продвигается в сторону реки. Вдруг мы замечаем что-то необычное. Как минимум пятая часть овец неистово катается по траве и трется шкурой о камни, выступающие над поверхностью земли, оставляя за собой беспорядочные клочья. Расчесанные, потерявшие шерсть бока покрыты страшными язвинами и эррозиями. Часть овец уже не способна чесаться, они просто медленно, трясущейся походкой, скрежеща зубами идут по полю, к месту последнего успокоения. Что же это за напасть такая, думали скотоводы, называя болезнь по ее главному проявлению – СКРЕПИ («почесуха»). Эта зараза не отступала столетиями, то и дело появляясь то там, то здесь, оставляя после себя разоренные семьи.
«Мозгоедки»
Настоящие ученые – не совсем обычные люди, знакомясь с их биографиями, часто поражаешься, каким же сумасшедшим вихрем крутилась шарманка судьбы.
Одна из ниточек нашего повествования начнется с жизнеописания Даниэля Карлтона Гайдусека (1923-2008). Представим себе молодого человека, ему 23 года, он только что получил степень магистра в Гарварде, с огромным воодушевлением едет работать в Калифорнийский технологический, да не абы с кем, а с самим Лайнусом Полингом (дважды Нобелевский лауреат). Спустя три года он принимает приглашение и занимает должность научного сотрудника факультета педиатрии и инфекционных болезней уже в своей альма-матер. Несмотря на столь успешную карьеру, что-то не ладится и не дает ему покоя. Не проработав и 3-х лет бросает все и уезжает сначала в Тегеран в институт Пастера, а через три года странным зигзагом через Гиндукуш, оказывается в медицинском институте Уолтера и Элизы Холл в Мельбурне. Дауншифтинг, не иначе.
Именно в Австралии, состоялось судьбоносное знакомство Даниэля Гайдусека с медицинским работником Винсентом Зигасом (1920-1983), который тесно общался с племенами Папуа Новой Гвинеи, оказывая им медицинскую помощь. Зигас рассказывает Даниэлю о неизвестной болезни, странные симптомы которой проявляются у единственного народа – Форе. Гайдусек в нетерпении бросается изучать язык аборигенов и спустя несколько месяцев Зигас привозит и представляет Гайдусека народности Форе. Почти год они живут среди дикого племени, отслеживая все привычки, обычаи. Наблюдают больных и проводят вскрытия погибших. [1]
Вот как они описывают последовательность развития столь заинтересовавшей их болезни в своей статье:
«… Человека настигает апатия и непреодолимая усталость. Спустя месяц или чуть больше начинаются характерные подергивания и подрагивания. Все более отчетливым и постоянным становится тремор конечностей, туловища и головы. Человек теряет способность передвигаться. В срок от года до двух лет наступает смерть. Члены племени Форе называют эту болезнь «Куру», что означает дрожь, порча. И считают, что причина кроется в сглазе шамана».
После проведения вскрытия, у многих погибших от болезни Зигас и Гайдусек обнаруживали превращения мозга в губчатую субстанцию. [2]
Длительное проживание внутри племени позволило Гайдусеку и Зигасу обнаружить причину развития болезни. Оказалось, что племя Форе практиковало каннибализм.
После смерти одного из старших членов рода его тело разделывали, вскрывали черепную коробку и съедали мозг, так как считалось, что поедание головного мозга представляет собой ритуал последних почестей умершему, а тот, кто съест мозг, приобретёт его мудрость, смелость и остальные благородные качества, которыми он владел. Обычно большую часть мозга съедали женщины и потому среди них число заболевших было выше.[3] С искоренением столь пагубного обычая практически полностью была побеждена и болезнь «куру».
За описание болезни «куру» в 1976 году Гайдусек получил Нобелевскую премию. И тут не ясны мотивы Нобелевского комитета, который обошел вниманием Винсента Зигаса. В своей Нобелевской лекции Гайдусек рассказывал про вирусную природу болезни «куру». Узнаем прав он был или нет чуть позже.
Больной, пораженный «куру».
«Успел»
Пока же перенесемся в Германию. Начало 20 века, психиатрическая клиника в Бреслау, кафедра под началом Алоиза Альцгеймера. На работу приходит молодой судовой врач, который решил стать неврологом. Пока он упорно постигает азы профессии ему удается обнаружить пациентов с никому дотоле неизвестным заболеванием. Исследования прерывает Первая мировая война, которая вернула доктора Ганса-Герхарда Крейтцфельда в состав военно – морского флота. Только в 1920 году, спустя 6 лет, он публикует описание болезни.
В описании обнаруживаем, что пациенты с высокой скоростью теряли память, переставали осознавать себя и через 8-12 месяцев после первых проявлений клинической картины умирали. В препаратах мозга, полученных от таких пациентов, были обнаружены характерные «губчатые структуры».
Стоит сказать, что ему несказанно повезло, опоздай он еще на полгода, и ветер времени развеял бы его имя в веках, так как спустя несколько месяцев выходит работа Альфонса Якоба с описанием той же самой болезни, которая обрела имя своих открывателей – болезнь Крейтцфельда – Якоба(БКЯ).
Что может быть общего между скрепи, куру и БКЯ? Именно такой вопрос начали задавать ученые к 50-м годам 20 века, ведь эти заболевания были так похожи длинным инкубационным периодом в 5 – 10 лет и неизменной печальной судьбой пораженного, будь то животное или человек. Причем повреждения прежде всего настигали мозг. Так и назвали эту группу заболеваний нейродегенерации с длительным инкубационным периодом.
Основная часть
Эксперименты
С развитием экспериментальных методов биохимии стало возможным наконец подступиться к этим патологиям. Неимоверно сложно было искать источник заражения при условии, что проявления болезни можно обнаружить только спустя годы, несмотря на трудности, попытки выяснить причины болезни не прекращались. В лабораториях искали способы упростить экспериментальную работу, сократив срок инкубационного периода до приемлемого.
Так Пэтисон и Гуили смогли передать болезнь от овечки к овечке с помощью бесклеточных фильтратов. Для начала лабораторных экспериментов оставался один шаг – передать болезнь от овцы к лабораторному животному. И его делает Чандлер в 1960 году, о чем пишет небольшую, но очень известную статью в 1961 году[4]. Ему удалось заразить лабораторную мышь с помощью вещества из мозга больного животного. Причем в последних исследованиях проявление заболевания пришлось ждать всего 7 месяцев. Стало удобно исследовать болезнь в лабораторных условиях.
Активизировались поиски инфицирующего агента. Установить его долгое время не удавалось. Сначала искали неизвестный вирус, похожий на герпес или энцефалит, но ничего не находили. Всех исследователей удивляло, что способность к заражению у этой субстанции, выделенной из мозга больных животных, сохранялась и после сильного длительного нагрева, и после обработки ацетиэтилениминам. Были поставлены эксперименты, в которых фильтрат подвергли обработке жестким УФ и ионизирующим излучением. Несмотря на это, фильтрат сохранил способность к заражению.[5] Стали закрадываться подозрения, что вирусы в этом случае ни при чем, ведь нуклеиновые кислоты (непременная составляющая любого вируса) при таком воздействии попросту разрушаются.
Гриффит в небольшой заметке на полторы страницы текста в 1967 году озвучивает еретическую мысль – инфекционный агент не содержит нуклеиновых кислот.[6] Это белок, который способен к самовоспроизводству в клетке. Именно с этой заметки началась новая эра.
Инфекционный белок
Эксперименты по исследованию скрепи все также оставались сложными и длительными. Только спустя 15 лет, Стенли Прузинер в калифорнийском университете Сан-Франциско выделил и описал агент, способный в чистом виде вызывать развитие болезни скрепи. Выяснилось, что это удивительное вещество устойчиво к нагреванию, сохраняет инфекционность после обработки различными повреждающими агентами, такими как: протеиназа К, мочевина, гуанидинхлорид, детергенты, SDS и нуклеазы — ферменты повреждающие ДНК, но было также обнаружено, что данный инфекционный агент чувствителен к ионизирующему излучению в присутствии кислорода, что характерно для гидрофобных белков имеющих большое сродство к липидам. [8] Прузинер придумал название для агента, вызывающего скрепи – «ПРИОН» (prion –proteinacious infectious particle). Прионный белок (Prione Protein PrP) был выделен чуть позже. Методы секвенирования в то время уже были развиты достаточно хорошо и быстро позволили установить первичную последовательность PrP. Все начали искать источник PrP. Статья в Nature от 1985 года, ознаменовавшая собой окончание поисков, поставила многих исследователей в тупик: матричная РНК (молекула – шаблон по которой потом синтезируются белки) необходимая для синтеза PrP обнаружилась в здоровом мозге. [7]
Это означало только одно — белок, ответственный за развитие болезни всегда присутствует в головном мозге, не зависимо от развития болезни. Позже выяснили, что ген, кодирующий PrP есть у всех млекопитающих, а также у птиц и рыб.
Расположение отдельных участков 20-й хромосомы человека с отметкой места нахождения гена, кодирующего PrPС.
Структура белка
Что же это за удивительный белок? Функция его и спустя 37 лет с момента обнаружения не выяснена (тут стоит сказать спасибо западной модели грантово-статейной науки). Известно, что этот белок связан с клеточной мембраной. И возможно отвечает за межклеточные взаимодействия в мозге.
Чтобы понять, как же обычный белок становится заразным необходимо обратиться к структуре белков. Первичная структура белка – это последовательность аминокислотных остатков. Эта последовательность и у нормального PrPC и у инфекционной формы PrPSc одинаковая.
Отличия удалось обнаружить на уровне вторичной и третичной пространственной структуры. У PrPC вторичная структура представлена 42% α-спиралей и 3% β-структур, а в тоже время PrPSc содержит 30% α-спиралей и 43% β-структур. Этот факт позволил предположить, что патологическая форма белка образуется при неправильном сворачивании аминокислотной последовательности в β-складчатые слои.
На изображении вверху аминокислотная последовательность PrP, с выделенными участками различных белковых структур h2,h3, Р3 – α спирали. Внизу показано превращение спиралей в β-складчатые слои. Изображение: By Olivia May, Ph.D
Прионная гипотеза
На основе накопленных данных в 1991 году Прузинер формирует «Прионную гипотезу», в которой постулирует следующее:
- инфекционным агентом является белок PrPSc,
- инфекционный агент PrPSc может реплицировать себя в отсутствие нуклеиновой кислоты,
- превращение белка из нормальной формы (PrPC) в инфекционную (PrPSc) происходит путем конформационного перехода,
- конформационный переход PrPC в PrPSc может происходить спонтанно, приводя к спорадическим формам прионных болезней. Он может быть вызван поступлением в организм патологической формы PrPSc извне (приобретенные формы прионных заболеваний),
- переход может произойти из-за мутаций в гене Prnp, способствующих образованию PrPSc из PrPC (наследственные формы прионных заболеваний). [9,10].
Таким образом у прионных болезней может быть причиной генетический дефект, заражение извне или их комбинация.
Несмотря на ярые атаки критиков этой теории, сейчас практически все соглашаются с тем, что Прузинер был прав, и этому есть значительный объем экспериментальных подтверждений. К примеру, если представить, что воспроизведение PrPSc после попадания в организм происходит путем передачи патологической конформации на PrPC, то организмы, лишенные PrPC, должны быть устойчивы к прионной инфекции. Такой эксперимент провели с использованием трансгенных мышей, гомозиготных по делеции гена Prnp (Prnp0/0). Введение растертой ткани мозга мышей, больных скрепи, трансгенным мышам Prnp0/0 не приводило к развитию болезни ввиду отсутствия нормального PrP. Более того, оказалось, что в отсутствие PrPC не происходит не только воспроизведения приона, но и повреждения нервной ткани.
Окончательное доказательство концепции прионов долгое время сдерживалось невозможностью получения значительного количества PrPres – формы PrPSc, образуемой in vitro, которая устойчива к частичному протеолизу и способна вызывать болезнь при введении экспериментальным животным. Недавно было показано, что фрагмент рекомбинантного PrP мыши, синтезированный в Escherichia coli, образует фибриллы in vitro, которые при введении трансгенным мышам, экспрессирующим этот же фрагмент PrP, приводят к развитию прионного заболевания. [13]
Также в недавнее время была разработана система циклической амплификации прионной формы белка PrP, с помощью которой возможно формирование значительного количества PrPres (искусственной патологической версии приона) in vitro. Это позволило получить и продемонстрировать инфекционность искусственно синтезированного приона.
Внимательный читатель заметит, что прионы, образующиеся в мозге овцы, вряд ли будут патогенны по отношению к человеку. И будут почти правы. Известно, что передача прионной инфекции между видами млекопитающих ограничена межвидовыми барьерами. Например, болезнь Крейцфельда-Якоба передается от человека человеку, и от человека шимпанзе; скрепи же передается среди овец и коз, но не передается шимпанзе. В то же время межвидовые барьеры не абсолютны. Межвидовые барьеры могут выражаться не столько в невозможности передачи инфекции животным отдаленного вида, сколько в удлинении инкубационного периода, а также в том, что заболевают не все, а какая-то часть экспериментально зараженных животных. Считается, что межвидовые барьеры вызваны различиями в первичной структуре PrP и модификациях у млекопитающих разных видов. Подтверждением этому послужили следующие наблюдения. Трансгенные мыши, экспрессирующие PrP хомяка, оказались высокочувствительны к заражению прионами хомяка в отличие от мышей дикого типа. Передача болезни Крейцфельда-Якоба от человека к мыши ограничена межвидовым барьером, однако трансгенные мыши, экспрессирующие PrP человека, подвержены заражению этой болезнью.
Также до сих пор встречаются трудности по заражению животных с помощью чистого прионного белка. Эти трудности можно легко объяснить.
Первая причина в том, что в клетках обычного организма белки подвергаются посттрансляционной модификации, которую достаточно трудно воспроизвести в экспериментальных условиях.
Вторая причина в том, что прион является белком мембранным и следует предполагать, что и структура его наиболее стабильна в условиях мембранно-подобного окружения, что и было показано недавними исследованиями.
В них было показано, что прионы в присутствии холестерина и фосфатидилэтаноламина гораздо легче образовывали патогенную форму и обладали гораздо большей инфекционностью.
Прионы — биологическое оружие
На этом можно было бы и закончить рассказывать про страшные прионы. Однако читатель законно спросит: « Причем же здесь биологическое оружие? Ведь чтобы произошло заражение необходимо, чтобы патогенная молекула приона попала в головной мозг. Не будем же мы сами себе делать трепанацию черепа».
На самом деле ситуация с возможными путями заражения оказалась гораздо хуже, чем можно было себе представить. В 1974 году был описан первый случай ятрогенного (из-за внешнего воздействия) заболевания болезнью Крейтцфельда-Якоба, обычно считавшейся генетической патологией.
Имеются описания 3 случаев передачи БКЯ в результате переливания крови от донора, у которого был диагностирован БКЯ во время вспышки этого заболевания в Великобритании [28]. От чего же произошла эта вспышка… Как обычно из-за жадности. БКЯ развилась у людей после употребления в пищу говядины, зараженной прионами.
В 1986 г. в Великобритании вспыхнула эпидемия заболевания прионнной болезнью у коров, также названной «коровьим бешенством», которая привела к гибели более чем 160 000 голов крупного рогатого скота [29]. Причиной было использование пищевых добавок мясокостной муки, когда из-за слабо контролируемых правил переработки побочных продуктов животного происхождения PrPSc от зараженных скрепи овец и другого крупного рогатого скота, попадал в корм для коров. Обычно в технологию получения такой муки после процессов тщательного измельчения исходного сырья включена обработка активными жирорастворителями, а также термообработка при температуре 130 оС. Однако в конце 70-х годов предприниматели, решив повысить питательную ценность мясокостной муки, снизили режим термообработки до 110 оС, а также уменьшили количество веществ, экстрагирующих жир. Именно эти изменения способствовали появлению и развитию эпидемии среди поголовья крупного рогатого скота.
Доказано, что эпидемия у коров привела к появлению нового типа БКЯ, получившего название «вариант БКЯ» [15]. Первые случаи вБКЯ были зарегистрированы в 1995 г., когда заболевание диагностировали у 2 британских подростков [16,17]. Из-за длительного инкубационного периода связь между заболеванием и зараженным мясом в Великобритании не была установлена до тех пор, пока заболеваемость у коров не переросла в эпидемию. Эпидемия была взята под контроль после массивного убоя скота и изменений в технологии производства, которые резко сократили загрязнение мяса компонентами нервной ткани. В Великобритании ежегодное число новых случаев вБКЯ, которое достигло пика в 2000 г., неуклонно снижается, и в 2013 г. был подтвержден только 1 случай заболевания [18].
У всех пациентов БКЯ развился после употребления в пищу мяса, полученного от заболевшего крупного рогатого скота. Но, несмотря на широкое распространение эпидемии, поразившей сотни тысяч голов крупного рогатого скота, относительно у немногих людей, которые употребляли в пищу мясо больных животных, развился БКЯ [33]. (вспомним про межвидовой барьер).
Инкубационный период (время между употреблением в пищу зараженной говядины и манифестацией симптомов) был длительным: большинство пациентов были заражены в конце 80-х годов, а пик заболеваемости пришелся на начало 2000-х, т. е. инкубационный период составил 11—12 лет. В последних диагностированных случаях инкубационный период достигал от 12 до более 20 лет [18.19].
Клинические проявления варианта БКЯ имеют отличия от других форм БКЯ. Болезнь настигает молодых людей в возрасте в среднем до 30 лет, ее начало характеризуется изменениями личности: больной утрачивает прежние интересы, начинает сторониться близких людей, у него развиваются тревожное состояние, бессонница, депрессия. Двигательные нарушения проявляются примерно через полгода после начала заболевания. Слабоумие наступает позднее, чем при классической форме, пациент осознает свое ухудшающееся состояние. Довольно быстро он теряет способность самообслуживания. Для вБКЯ типичны не только начало в более молодом возрасте, но и средняя выживаемость, превышающая 14 мес [18,19]. Вероятно, что различия в выживаемости между классической БКЯ и ее вариантом отчасти связаны с молодым возрастом пациентов.
Вот так, сама природа нам продемонстрировала возможность использования прионов как оружия с отсроченным сроком воздействия.
К глубочайшему сожалению в 2011 году во время экспериментальных работ по изучению болезни Крейтцфельда – Якоба на мышах была показана возможность воздушно – капельного заражения аэрозолями, содержащими прионные частицы.
Прионы – идеальное биологическое оружие?
В чем же заключаются главные достоинства:
- Болезнь проявляется в отсроченном периоде, у злоумышленника есть время чтобы заразить как можно большее число людей. При этом все окружающие будут находится в абсолютном неведении.
- Можно прекратить заражение, также незаметно, как и начать его. Найти следы и источник заражения спустя 5-7 лет будет невероятно трудно. Тем более нужно будет знать, что искать.
- Высокая инфекционность прионов. При неудачном стечении обстоятельств для заражения теоретически достаточно одной молекулы неправильно свернутого белка, если эта молекула проникнет через гематоэнцефалический барьер и свяжется с обычной версией прионного белка на поверхности нейрона
- Белок в обычных условиях присутствует в животном биоматериале и невозможно простыми способами отличить обычный прион от патогенного.
- Прионы устойчивы во внешней среде, не разрушаются при стерилизации. Очень трудно расщепляются протеиназами. Способны связываться с частицами почвы, оставаясь стабильными долгое время.
- Нет достаточно надежного диагностического теста для ранней стадии развития заболевания.
- Против прионов нет вакцины или иного лекарства. Человек, заразившийся прионами обречен [9]
Помогаем террористам осуществить хитрый замысел
Прионы — это идеальная технология для террора. Существует и хорошо описана технология синтеза патогенных форм прионных белков[13].
Даже если представить, что обычному террористу сложно будет организовать биохимическую лабораторию, то бесчисленные стада животных никто не мешает использовать для получения большого количества мозгового вещества, зараженного БКЯ.
Никто и ничто не помешает террористам начать массовый синтез прионных белков и их добавление к сухому молоку, детским молочным смесям, мясному фаршу, мясо-костным субпродуктам, соевому шроту или любой другой субстанции, завод по производству которой окажется в зоне их досягаемости.
Если же представить, что в руки террористов попадет насильственным способом или по финансовым, идейным, иным соображениям талантливый биохимик, то никто не помешает ему синтезировать липидно-белковый аэрозоль с прионными частицами. Потом распылять ничем не определяемый аэрозоль в системах вентиляции. Этот способ более страшный, чем через пищу, так-как у носоглоточного узла есть тесная связь с мозгом и вероятность заражения увеличивается многократно.
Представим себе последствия заражения. Спустя 3-7, а может и все 15 лет на неограниченной территории начинается массовое развитие прионной болезни мозга. Паника, ужас, страх, разрушение. Целые города людей — зомби, чей мозг в буквальном смысле превращается в губку. Нет лекарства, нет надежды, только ужас неотвратимой скорой гибели.
Заключение
Использование такого оружия – только дело времени. Поэтому уже сейчас нужно предпринимать ряд шагов:
- Проводить исследования по созданию надежных систем обнаружения патологических прионов в продуктах питания, воде, воздухе и делать этот тест обязательным к использованию на всей территории земного шара. Внедрять системы обнаружения прионных белков.
- Искать возможность диагностики у человека появления патологических прионов. Тут есть радостные вести, что разработан высокочувствительный метод обнаружения патологических прионов. [12]
- Искать способ излечения человека. Что представляется невероятно трудным, несмотря на многообещающие достижения с антиприонными антителами, способными проникать через гематоэнцефалический барьер.
- Отслеживать на уровне спецслужб массовые случаи заражения животных прионными заболеваниями.
Новогоднее пожелание
Пожелаю никогда не встретить ни одной молекулы PrPSC!
Сам пока поищу возможность достать высокочувствительную диагностическую тест — систему, чтобы определить, не успели ли нас уже заразить…
Ссылки на источники1. www.nobelprize.org/prizes/medicine/1976/gajdusek/biographical2. Gajdusek, D. C.; Zigas, V. (1957-11-14). «Degenerative Disease of the Central Nervous System in New Guinea». New England Journal of Medicine. 257 (20): 974–978.
3. Hussain Khan, C.G. Bio-medical Paradigm // Bio-social issues in health. General editor, R.K. Pathak. New Delhi: Northern Book Centre, 2008. — p. 15
4. Chandler R.L. (1961) Lancet, 1,1378–1379
5. Alper T., Cramp W.A., Haig D.A., and Clarke M.C. (1967) Nature, 214, 764–766.
6. Griffith J.S. (1967) Nature, 215,1043–1044.
7. Chesebro B., Race R., Wehrly K., Nishio J., Bloom M., Lechner D., Bergstrom S., Robbins K., Mayer L., Keith J.M., et al. (1985) Nature,315, 331–333.
8. Prusiner S.B. (1982) Science, 216,136–144
9. Prusiner S.B. (1991) Science, 252,1515–1522
10. Prusiner S.B. (1993) Proc. Natl. Acad. Sci. USA Vol. 90, pp. 10962-10966, December 1993 Biochemistry
11. Saima Zafar et al., Handbook of Clinical Neurology, Vol. 165, 2019 (3rd series)
12. Serena Singh, Mari L. DeMarco JALM, January 2020
13. Nature Communications (2018) Chae Kim, Xiangzhu Xiao, Shugui Chen, Tracy Haldiman, Vitautas Smirnovas, Diane Kofskey, Miriam Warren, Krystyna Surewicz, Nicholas R. Maurer, Qingzhong Kong, Witold Surewicz & Jiri G. Safar Artificial strain of human prions created in vitro volume 9, Article number: 2166
14. Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Possible transmission of variant Creutzfeldta Jakob disease by blood transfusion. Lancet. 2004;363:417-421.
15. Collinge J. Human prion diseases and bovine spongiform encephalopathy (BSE). Hum Mol Genet. 1997;6(10):1699-1705
16. Bateman D, Hilton D, Love S, Zeidler M, Beck J, Collinge J. Sporadic Creutzfeldt Jakob disease in a 18-year-old in the UK. Lancet. 1995; 346(8983):1155-1156.
17. Britton TC, al-Sarraj S, Shaw C, Campbell T, Collinge J. Sporadic Creutzfeldt—Jakob disease in a 16-year-old in the UK. Lancet. 1995; 346(8983): 1155.
18. Soomro S, Mohan Ch. Biomarkers for sporadic Creutzfeldt Jakob disease. Annals of Clinical and Translational Neurology. 2016;3(6):465-472.
19. Imran M, Mahmood S. An overview of human prion diseases. Virol J. 2011;8:559.
что это такое, какие болезни вызывают
Инфекционные болезни могут развиваться не только из-за бактерий, вирусов и других хорошо известных врачам микроорганизмов - это ошеломляющее заявление в медицинской среде было сделано после того, как в 1982 году профессор неврологии и биохимии Стэнли Прузинер (США) обнаружил белковые соединения, способные вызывать заболевания. Открытие белков-прионов было настоящим прорывом в медицине, доказательством чему стало получение учёным Нобелевской премии в 1997 году.
Прионы: биологическая сущность, свойства, среда обитания таинственных молекул
До недавнего времени исследователи считали, что в составе любой «живой» субстанции должны быть молекулы ДНК или РНК — нуклеиновых кислот, обусловливающих способность вирусов, бактерий, грибов и прочих организмов размножаться. Однако открытие прионов полностью трансформировало это представление. Устойчивость к высоким температурам, к различным видам излучений, действию нуклеаз (ферментов, способных расщеплять нуклеиновые кислоты), отсутствие роста на питательных средах – такими необычными свойствами обладал ранее не известный возбудитель.
Белковые соединения с определённой конфигурацией, способные трансформироваться в патогенные и вызывать нейродегенеративные процессы в организме, были названы прионами. Термин «прион» (prion) предложил Стэнли Прузинер. Термин происходит от фрагментов английских слов protein (белок) и infection (инфекция). Прионы способны размножаться. Этот процесс более продолжителен по времени, чем размножение патогенных микроорганизмов, поэтому от момента попадания прионов в организм до клинических проявлений болезни может пройти несколько месяцев или лет.
Молекула приона в «нормальной» форме имеется на поверхности нервных клеток у каждого человека. Обычные молекулы белка, вступая в контакт с патологическими, сами превращаются в них, изменяя при этом собственную пространственную структуру. Что является пусковым механизмом подобной трансформации, до конца не известно. Из этого следует, что прион, выступая в роли инфекционного агента, заражает нормальные молекулы, вызывая «молекулярную эпидемию».
Токсичные белковые бляшки на клетке приводят к её гибели, а на месте погибшей клетки образуется пустота, которая заполняется жидкостью. Количество пустот в головном мозге с течением времени будет увеличиваться, пока он не превратится в «губку».
Как можно заразиться прионами?
На сегодняшний день выделяют следующие основные пути заражения инфекционным белком-прионом:
1. Трансмиссивный. В этом случае молекулы белка передаются от одного вида млекопитающего к другому — например, от инфицированной коровы или овцы человеку. Заражение происходит при употреблении в пищу мяса или молока заражённого животного, либо использовании его тканей (роговицы, препаратов крови и т.п.), применении во время оперативных вмешательств биологического шовного материала.
2. Наследственный. Заболевание развивается на фоне генетической мутации, затрагивающей область 20-й хромосомы. Несмотря на слабую изученность функционирования этого участка генома, достоверно известно его участие в синтезе нормального прионного белка. В случае генных мутаций вместо здорового приона образуется патологический, а это приводит к развитию болезней.
3. Спорадический. При этом аномальный белок появляется в организме спонтанно, без видимых причин.
Вне зависимости от способа появления аномальный белок может стать причиной заражения других людей.
Прионные заболевания: особенности течения, лечения, прогноз
Отличительной особенностью болезней, вызываемых прионами, является длительный инкубационный период - от 2-3 месяцев до нескольких десятилетий. Подавляющее большинство прионных заболеваний человека являются спорадическими и имеют семейный характер наследования.
Куру, синдром Герстманна-Штреусслера-Шейнкера, болезнь Крейтцфельдта-Якоба, скрэпи – прионы вызывают заболевания, сопровождаемые поражением центральной нервной системы. Для них характерны такие признаки как деменция (слабоумие), зрительные и мозжечковые нарушения. При этом у больного могут отмечаться двигательные расстройства, бессонница, галлюцинации, нарушение речи.
К сожалению, эффективных методов лечения прионных болезней на сегодняшний день нет, хотя учёные пытаются предотвращать переход нормального белка в аномальный. Пациентам назначается симптоматическая терапия с использованием противосудорожных средств для облегчения страданий. Прогноз пока неутешителен, так как все вышеперечисленные заболевания завершаются летальным исходом.
Перспективы
Недостаточная изученность проблемы прионов и прионных болезней способствует углублению исследований в этой области — учёные занимаются активным поиском средств борьбы с патогенными белками. Актуальность этого вопроса растёт в связи с возможностью возникновения «прионной эпидемии», например, из-за приёма лекарственных средств животного происхождения.
Раскрытие загадочных явлений, которыми окутаны прионы, возможно, поможет в понимании ряда серьёзных биомедицинских проблем человечества.
Севиля Ибраимова
Редакция рекомендует:
Не антибиотиком единым: «киллеры» бактерий – бактериофаги
Риск на грани. Как открыли хеликобактер пилори?
Прионы в мозге человека: болезнь которая убивает | Онлайн-доктор для всей семьи
Прионная болезнь у взрослых вызывается возбудителями – белками-прионами. Это белки с аномальным строением, которые остановились на третичной структуре развития. Попадая в организм, они не вызывают воспалительных реакций или ответов иммунитета, даже если циркулируют в крови. Любимое место скопления прионов – головной мозг. Симптомы прионной инфекции у взрослых напоминают неврологические патологии (эпилепсии, нарушения работы мозжечка). Лечения прионной инфекции не существует, но случаев ее развития в странах СНГ немного, до 3-4%. Есть только неспецифическая профилактика не наследственных форм заболевания.
Механизм заражения и течения болезни
Опасность прионной болезни у взрослых заключается в поражении мозга и мозговых оболочек. Белки вызывают спонгиозные или губчатые энцефалопатии, поражая и белое и серое вещество. Анатомически и морфологически в тканях мозга происходят следующие изменения:
 Размягчение белого вещества, образование дырок или полостей в мозге. Формируется губчатая, неоднородная структура.
Размягчение белого вещества, образование дырок или полостей в мозге. Формируется губчатая, неоднородная структура.- Происходит гибель нейронов.
- Вместо получившихся дырок мозговое вещество выполняется соединительной тканью.
- Происходит атрофия мозга.
- На месте скопления прионов развиваются амилоидные бляшки.
Типичных признаков инфекции во время прионной болезни нет. Процессы дегенерации и атрофии головного мозга происходят без воспаления. Организм не распознает эти белки как чужеродные, не запускается каскад иммунных реакций.
Опасность прионов заключается в том, что попадая в головной мозг или места других скоплений белков, они способны вызывать перерождение нормальных белковых структур. То есть, вокруг попавшего приона появляется целый конгломерат новых мутантных белков. В дальнейшем очаги с прионами превращаются в амилоидные бляшки. Прионная инфекция у взрослых отличается от амилоидоза тем, что развивается быстрее, степень поражения и изменения тканей сильнее. Соответственно функция пораженного органа теряется в течение 5–7 лет.
Заболевания, которые провоцирует прионная инфекция у взрослых
Ряд патологий, причинами которых является прионная болезнь у взрослых:
 Болезнь Герстманна—Штреусслера—Шайнкера. Это наследственная патология, которая передается по аутосомно-доминантному типу. У пациента с 30-40 лет начинаются нарушения в опорно-двигательном аппарате, развивается тремор рук, нарушается координация движений. Манифестирование (проявления болезни) может произойти и позже, в 50-60 лет. Неизвестно, что становится пусковым рычагом инфекции. На поздних этапах заболевания возникает слабоумие.
Болезнь Герстманна—Штреусслера—Шайнкера. Это наследственная патология, которая передается по аутосомно-доминантному типу. У пациента с 30-40 лет начинаются нарушения в опорно-двигательном аппарате, развивается тремор рук, нарушается координация движений. Манифестирование (проявления болезни) может произойти и позже, в 50-60 лет. Неизвестно, что становится пусковым рычагом инфекции. На поздних этапах заболевания возникает слабоумие.- Куру. Эта патология была обнаружена в племенах Папуа-Новой Гвинее. Там был распространен каннибализм. Процесс заражения происходил при поедании мозга врагов или соплеменников. Заболевание сопровождалось сильным тремором, подергиванием головы и улыбкой на лице (прионная болезнь прозвали «смеющейся смертью»).
- Заболевание Крейтцфельда-Якоба. Поражает ЦНС и проявляется постепенными нарушениями координации движений, слабоумием, расстройством памяти и ориентации в происходящем. Возможны эпилептоидные припадки.
- Фатальная семейная бессонница (инсомния). Это заболевание с наследственной передачей прионного белка по аутосомно-доминантному типу. Пациент не может заснуть, у него повышается артериальное давление, происходят расстройства памяти, появляются галлюцинации и страхи. Время на сон сокращается постепенно ,доходя до 10-15 минут за сутки. Пациент может уснуть днем, сон поверхностный, до получаса. Человек со временем перестает реагировать на происходящее и погибает.
- Хроническая прогрессирующая спонгиозная энцефалопатия детского возраста (болезнь Альпера). Заболевание передается по аутосомно-рецессивному типу и проявляется у детей до 18 лет. Поражения нервной системы характеризуются нарушением зрения, эпилептоидными припадками с конвульсиями и спазмами мышц. В 30-40% случаев развивается инсульт. Характерно поражение печени (хронический гепатит с переходом в цирроз).
Пути заражения прионами
Существуют три способа заражения прионной инфекцией у взрослых или прионными белками:
 Наследственный. Происходит мутация генов в области 20 хромосомы. В норме участок 20 хромосомы отвечает за формирование нормального прионного белка. Если происходят нарушения в генной структуре, то возникают прионные заболевания. Механизм мутации, а также полные функции данного гена не изучены.
Наследственный. Происходит мутация генов в области 20 хромосомы. В норме участок 20 хромосомы отвечает за формирование нормального прионного белка. Если происходят нарушения в генной структуре, то возникают прионные заболевания. Механизм мутации, а также полные функции данного гена не изучены.- Трансмиссивный или контактный. Прионы передаются от животного к животному или человеку. Прионная болезнь у взрослых развивается после употребления мяса зараженного животного, молока. Также белки передаются, если использовать ткани зараженного животного или человека (в том числе при переливании крови, пересадки биологических тканей). Количество прионов отличается от вида ткани. Наиболее опасной считается мозговая ткань. Поэтому прионные болезни были распространены на африканском континенте, в Индонезии по причине каннибализма и употребления в ритуалах мозга врага. На втором месте по вирулентности находятся препараты крови.
Возникают и спорадические (единичные) случаи заражения прионами. Причины этого явления не выяснены.
ВНИМАНИЕ! Ни один способ обработки пищи (зараженного мяса) не может защитить от попадания белков-прионов в организм. Эти аномальные белки плохо денатурируются (теряют структуру) и поддаются деструкции при любой кулинарной обработке.
Лечение прионовых заболеваний
Прионные болезни неизлечимы на данный момент. Врачи не могут повлиять на механизмы развития болезней и даже затормозить их. Продолжительность жизни человека, пораженного прионной инфекцией, зависит от собственных реакций организма.
ВНИМАНИЕ! Средняя продолжительность жизни взрослого пациента с прионной инфекцией составляет от 5 до 7 лет.
Единственное, что могут предложить врачи – кратковременная симптоматическая терапия. По необходимости назначают противосудорожные препараты (если есть эпилептические приступы), гемодиализ при поражении почек при прионной инфекции.
Смертность от прионной инфекции у взрослых – 100%.
Как уберечься от прионных инфекций?
 От наследственных форм прионных инфекций невозможно уберечься. На территории стран СНГ не существует лабораторий, которые проводят диагностику в пренатальном периоде и определяют возможность развития прионных инфекций у детей. Поэтому исключить вероятность такого диагноза в период беременности женщины нельзя.
От наследственных форм прионных инфекций невозможно уберечься. На территории стран СНГ не существует лабораторий, которые проводят диагностику в пренатальном периоде и определяют возможность развития прионных инфекций у детей. Поэтому исключить вероятность такого диагноза в период беременности женщины нельзя.
Чтобы обезопасить себя от алиментарного механизма передачи прионной инфекции, необходимо употреблять в пище мясные продукты только от проверенного производителя. Не рекомендуется употреблять лекарственные средства с добавлением препаратов крови человека.
 Загрузка…
Загрузка…Прионы: болезни и лечение — vechnayamolodost.ru
Молекулярный биолог Байрон Кои о структуре прионов, прионных болезнях, их причинах и лечении
ПостНаука
Прионы составляют отдельный класс инфекционных агентов; они имеют белковую основу и не содержат генома, состоящего из нуклеиновых кислот. Концепция существования прионов включает в себя идею о новой, белковой наследственности – измененных формах белков в организме-хозяине, которые при переносе в новый организм могут вызвать в получателе изменение фенотипа. Большинство прионов являются патогенами, но по сравнению с другими классами патогенов (например, вирусами, бактериями, грибами, паразитами) они уникальны тем, что распространяются внутри и между хозяевами без переноса или репликации собственных ДНК или РНК.
Прионы, как правило, – это повторно свернутые и агрегированные белки, которые распространяются, внедряясь в организм-хозяина и стимулируя в нем рефолдинг соответствующей нормальной формы белка. Прионный агрегат растет, а затем каким-то образом фрагментируется, чтобы сгенерировать больше прионных агрегатов. Во многих отношениях это похоже на лед-девять Курта Воннегута: рост прионов аналогичен росту кристаллов, а сами прионы часто описывают как затравку по аналогии с затравочными кристаллами. Таким образом, прионам не нужно переносить свой генетический код, но организм-хозяин должен создать нормальный белок, из которого состоят патогенные прионы. Многие белки (если не большинство) могут перегруппироваться и/или собираться в упорядоченные агрегаты, которые в определенных естественных или экспериментальных условиях – в тканях или пробирке – могут расти, однако не все такие белковые агрегаты являются патогенными прионами. Термин «прион» означает, что на биологическом уровне структура повторно свернутого белка может распространяться между хозяевами или по меньшей мере из клетки в клетку внутри многоклеточного организма-хозяина. Недавно звучало предложение собрать под зонтичным термином «прионы» все состояния белка, которые способствуют его росту в форме мультимерных скоплений, но это расширенное использование термина нарушает лежащую в его основе идею передаваемости и не позволяет выделить прионы из многих других клеточных структур, которые могут расти, но не имеют тенденции распространяться на другие клетки и организмы.
История исследований
Прототипом прионной болезни было трансмиссивное нейродегенеративное заболевание овец – таинственная смертоносная скрейпи (или почесуха овец). Ранние исследования показали, что агент скрейпи необычайно устойчив к лечению, которое нейтрализует другие патогены, и может годами оставаться на пастбищах. То, что агент скрейпи проявляет устойчивость, в частности, к радиации, привело к тому, что в 1960-х годах Дж. С. Гриффит и Тиквах Альпер предположили, что он представляет собой новый класс патогенов, который не имеет собственного нуклеинового генома и может быть аномальной самовоспроизводящейся формой белка или мембраны. Между тем описания патологии мозга, вызванной человеческой болезнью куру в Папуа – Новой Гвинее, которые выполнил Карлтон Гайдушек, привели Уильяма Хэдлоу к мысли, что куру похожа на скрейпи овец, и Хэдлоу порекомендовал, чтобы куру испытали на передаваемость от людей к другим приматам. Гайдушек успешно проделал эту работу и показал, что люди племени форе заболевали куру во время ритуальных каннибалистских праздников. Яркой особенностью куру и других прионных заболеваний, часто скрывавшей их причины, является длительный инкубационный период между заражением и появлением клинических признаков, который у людей может превышать четыре десятилетия.
Лимфатические узлы здоровой (а) и инфицированной (b) овцы – окрашивание при помощи антител четко демонстрирует признаки прионов болезни скрейпи в тканях инфицированной овцы / wikipedia.org
В 1980-х годах Стэнли Прузинер придумал для этих агентов термин «прион» и первым определил специфический белок, который является основным компонентом прионов скрейпи, – прионный белок (PrP). Гомологи того же белка были обнаружены у многих видов млекопитающих, в том числе и у людей, а аномальные агрегаты PrP выявлены в других трансмиссивных нейродегенеративных заболеваниях людей и животных, подобных скрейпи. Эти заболевания теперь известны как прионные заболевания, или трансмиссивные губчатые энцефалопатии. Стэнли Прузинер, Чарльз Вайсман и другие исследователи показали, что PrP является важным фактором восприимчивости прионных заболеваний.
Моя лаборатория в сотрудничестве с Питером Лэнсбери показала, что связанные с заболеваниями формы PrP сами могут вызвать трансформацию нормальных молекул PrP в аномальные формы. В этих реакциях превращения мы выявили поразительные биохимические особенности, которые помогли объяснить характеристики известных штаммов прионов и барьеры их передачи между разными видами. Однако, чтобы однозначно доказать, что прионы состоят из повторно свернутых агрегатов PrP и им не нужны специфически-прионные нуклеиновые кислоты, потребовалось разработать методы непрерывной бесклеточной амплификации прионов или реакции образования прионов de novo. Они первоначально были созданы лабораториями Сото, Супаттапоне и Прузинера в 2000-х годах; до того времени было трудно полностью исключить вероятность того, что эти заболевания вызваны неопознанными вирусами.
Хотя слово «прион» впервые было применено к описанным выше трансмиссивным губчатым энцефалопатиям, первые однозначные доказательства того, что в биологии существуют инфекционные белки, были получены, когда Рид Уикнер в 1994 году понял, что некоторые необъяснимые эпигенетические элементы в дрожжах – это прионы. Эти прионы состояли не из гомолога PrP, а из совершенно особенных белков дрожжей. Относительная простота и мощь биологии и генетики дрожжей позволили Уикнеру и другим исследователям ясно продемонстрировать ряд основополагающих принципов прионной биологии и структуры, которые было гораздо сложнее выявить на прионах млекопитающих.
Методы исследования
К сожалению, многие из стандартных методов, на которых долгое время базировались исследования обычных патогенов, – генетика патогенов, серология, рентгеноструктурный анализ, спектроскопия ядерного магнитного резонанса (ЯМР) – чрезвычайно трудно применить к прионам. Без каких-либо специфических патогенных генов, которые можно было бы секвенировать или подвергнуть мутации, многие стандартные генетические и обратные генетические подходы к выявлению структуры и функции патогенов не работают. Поскольку прионы состоят из белков организма-хозяина, иммунный ответ хозяина на патоген очень мал; таким образом, провести простое серологическое обнаружение прионных инфекций, основанное на взаимодействии с антителами, очень сложно. Кроме того, прионы млекопитающих, как правило, плотно упакованы, сильно гликозилированы и связаны с другими молекулами организма-хозяина, и поэтому даже специфические прионные конформационные эпитопы (поверхности, распознаваемые антителами) на агрегатах PrP трудно обнаружить и использовать. Все попытки определить трехмерные структуры прионов на протяжении уже долгого времени заходят в тупик, так как очищенные прионы имеют агрегированный, но некристаллический характер.
В течение многих лет единственным способом обнаружения и анализа прионов млекопитающих был биоанализ животных, который даже на самых быстрых моделях – грызунах – длился от нескольких месяцев до одного года. В конкретном организме разные штаммы обычно можно различить по периодам инкубации, невропатологическим паттернам и биохимическим признакам связанных с болезнью отложений PrP или прионов.
К счастью, в последнее время были разработаны мощные бесклеточные амплификационные анализы прионов, такие как циклическая амплификация прионной формы белка (PMCA), вибрационно-индуцированный конверсионный анализ в режиме реального времени (RT-QuIC) и анализ клеток скрейпи. Эти методы основаны на присущем прионам механизме репликации. И PMCA, и RT-QuIC чрезвычайно чувствительны: они могут усилить присутствие прионов в триллион раз, почти до точки обнаружения нескольких прионных частиц. Реакции PMCA распространяют инфекцию прионов, тем самым отражая и освещая многие аспекты прионной биологии, в то время как анализы RT-QuIC, как правило, не распространяют полностью инфекционные прионы, но обеспечивают более быстрые, более практичные и более высокопроизводительные методы их обнаружения, и, таким образом, они стали самыми современными инструментами в диагностике прионных заболеваний. Как PMCA, так и RT-QuIC в некоторых случаях помогают различать важные штаммы прионов у определенных видов организмов-хозяев.
В выявлении базовой структуры прионов наблюдается медленный прогресс. При помощи полупроводниковых ЯМР-исследований была обнаружена молекулярная архитектура некоторых прионов грибов и прионоподобных фибриллярных структур PrP млекопитающих. Электронная кристаллография, дифракция волокон и криоэлектронные микроскопические исследования помогли описать ключевые структурные ограничения прионов млекопитающих, но применение этих и, возможно, других структурных биологических методов еще нужно улучшить.
Структура и воспроизведение прионов
Разобраться в структуре и механизмах репликации прионов млекопитающих, по крайней мере на молекулярном уровне, крайне сложно. Сначала нужно объяснить, как неправильно свернутые белки могут распространяться в роли патогенов, не перенося своего собственного нуклеинового генома. Затем следует также объяснить, как белки с единой последовательностью аминокислот, такие как PrP того или иного животного-хозяина, могут образовывать разные штаммы прионов, которые исправно распространяются и вызывают различные фенотипы болезни без генетических мутаций, объясняющих вариации штаммов в обычных патогенах.
Множество исследований указывает на то, что прионы млекопитающих – это упорядоченные скопления нескольких молекул PrP, плотно упакованных и часто фибриллярных или нитевидных. Молекулы PrP (мономеры) в прионах по сравнению с нормальными свободными молекулами PrP пересвернуты практически полностью. Когда правильные молекулы PrP включаются в растущие прионные агрегаты, эти агрегаты вызывают их рефолдинг, причем прионы действуют как штамм-специфические шаблоны или затравки, которые каким-то образом придают свои собственные аберрантные формы каждой входящей молекуле, контролируя стабильную репликацию своего штамма.
За рамками этого грубого описания детали структуры и распространения прионов на молекулярном уровне остаются неясными. Также нерешенным остается вопрос о том, как прионы распространяются за пределы исходного места заражения в организме-хозяине. Существующие данные свидетельствуют о том, что наиболее эффективная межклеточная передача прионов связана с мембранозными структурами, такими как экзосомы или туннелирующие нанотрубки, – скорее всего, потому, что прионы обычно связаны с мембранами липидными якорями; однако возможность этих мембранных структур способствовать распространению прионов in vivo еще предстоит определить. Очень важно понять механизмы распространения прионов, поскольку способности различных неправильно свернутых белковых агрегатов распространяться внутри и между клетками, тканями и индивидами определяют то, действуют ли они как инфекционные патогены или являются относительно безобидными сбоями белкового метаболизма.
Прионные болезни
Многие виды млекопитающих, включая людей, низших приматов, крупный рогатый скот, овец, коз, оленей, лосей, кошек, норок, грызунов и различных экзотических копытных, восприимчивы к прионным заболеваниям PrP. Но такими являются не все виды: собаки и лошади, судя по всему, представляют собой заметные исключения. Разные виды обычно экспрессируют несколько разные нормальные молекулы PrP, и различия в аминокислотной последовательности PrP могут сильно влиять на восприимчивость хозяина к входящим прионным инфекциям. Например, люди, как известно, до некоторой степени восприимчивы к губчатой энцефалопатии крупного рогатого скота (ГЭКРС), но, по-видимому, устойчивы к скрейпи овец и, насколько нам известно, хронической изнуряющей болезни оленей. По какой-то причине лесные полевки и беличьи обезьяны необычайно восприимчивы к широкому спектру прионных инфекций других видов.
Механизмы, с помощью которых прионные инфекции вызывают нейродегенеративные заболевания, нам пока неизвестны. Агрегаты различных прионных штаммов в организмах-хозяевах разных видов могут накапливаться преимущественно в разных областях центральной нервной системы и вызывают ряд невропатологических расстройств. Очевидно, что конечным эффектом по крайней мере частичного повреждения является сбой в работе нейронов и их потеря, что вызывает множество клинических симптомов и приводит к летальному исходу. Известно, что ряд нейрофизиологических процессов и путей нарушается, но многое еще предстоит определить относительно того, связаны ли такие нарушения с прямой или косвенной токсичностью прионов и в какой степени та или иная недостаточность или комбинация недостаточностей наиболее ответственна за кончину больного.
У людей причины прионных заболеваний могут быть генетическими (из-за специфических мутаций гена PrP), приобретенными (вызванными заражением – например, воздействием куру, ГЭКРС или другим содержащим прионы материалом) или спорадическими (неизвестного происхождения; обычно предполагается, что они обусловлены спонтанным образованием прионов у конкретного индивидуума). Подавляющее большинство прионных заболеваний человека являются спорадическими, и среди них наиболее распространена спорадическая болезнь Крейтцфельдта – Якоба (sCJD), заболеваемость которой в год во всем мире составляет примерно один случай на миллион населения. Ряд различных мутаций в гене PrP может вызывать множество семейных прионных заболеваний, при этом некоторые мутации являются полностью пенетрантными (всегда вызывающими болезнь у носителей мутации), а другие – менее пенетрантными. Клинические симптомы и прогрессирование болезни могут заметно различаться в разных организмах-хозяевах и при разных типах прионных заболеваний, но могут включать деменцию, расстройство координации, бессонницу, галлюцинации, жесткость мышц, спутанность сознания, усталость и затрудненность речи.
Существуют также важные прионные заболевания животных. ГЭКРС возникла как масштабная эпидемия крупного рогатого скота в связи с тем, что можно было бы назвать «сельскохозяйственным каннибализмом» в 1990-х годах. Потребление ГЭКРС – зараженной говядины – вызвало тогда почти двести случаев варианта болезни Крейтцфельдта – Якоба у людей, однако превентивные меры почти ликвидировали ГЭКРС и предотвратили появление новых случаев. Хроническая изнуряющая болезнь оленей прокатывается по Северной Америке с угрожающей скоростью, причем случаи болезни также возникают в Южной Корее и Норвегии. Скрейпи – это постоянная проблема с овцами и козами во многих частях мира.
Диагностика и лечение прионных болезней
В последнее время были достигнуты значительные успехи в том, чтобы точно и относительно неинвазивно диагностировать прионные заболевания человека у живых пациентов на основе новых прион-специфических тестов мазков из носа, спинномозговой жидкости, крови, мочи или кожи. Например, RT-QuIC-тестирование спинномозговой жидкости и/или материалов назальной щеточной биопсии может достигать 100% точности при диагностике спорадической болезни Крейтцфельдта – Якоба. Эти тесты выгодны потому, что измеряют возбудителей прионной болезни, но они еще не полностью проверены и не рекомендованы официально такими организациями, как ВОЗ. В остальном диагностика спорадических прионных заболеваний у людей зависит в первую очередь от совокупности клинических признаков, результатов сканирования мозга, электроэнцефалограмм и других биомаркеров, которые вместе могут иметь высокую диагностическую чувствительность, но не полностью специфичны для прионных болезней.
Несмотря на описанные выше недавние успехи в разработке новых прионных тестов, действующие руководящие принципы таковы: для окончательной диагностики спорадического или приобретенного прионного заболевания необходимо невропатологическое исследование тканей головного мозга, полученных в результате биопсии (что редко) или аутопсии. Полагаю, в скором времени эти рекомендации будут изменены, в них будут включены новые, менее инвазивные прижизненные тесты для выявления прионов. К сожалению, несмотря на то, что этот прогресс в раннем диагностировании прионных болезней должен улучшить перспективы разработки и применения терапевтических средств, в настоящее время доступных методов лечения, которые доказали бы свою эффективность в клинических испытаниях, не существует.
Открытые вопросы и будущие направления исследований
В области прионных заболеваний млекопитающих остаются открытыми следующие ключевые вопросы: «Какова самораспространяющаяся структура прионов и как она варьируется в зависимости от штамма приона?», «Как деятельность прионов приводит к повреждениям мозга?», «Как мы можем предотвратить эти повреждения или восстановить их при лечении прионных заболеваний?», «Каковы наиболее актуальные механизмы передачи прионных заболеваний у людей и животных?», «Какие прионные заболевания животных (помимо ГЭКРС), если такие существуют, имеют зоонозный потенциал, то есть могут вызывать болезнь у людей?», «До какой степени другие патогенные, неправильно свернутые белки, которые также могут выступать в качестве затравки, ведут себя как основанные на PrP прионы в своей способности распространяться внутри или между людьми, вызывая болезнь?»
Действительно, последний вопрос представляет собой важный рубеж в изучении многих заболеваний, связанных с неправильным образованием белков, особенно тех, которые связаны с патогенным накоплением аномальных фибриллярных белковых отложений (например, амилоидных фибрилл и бляшек). Эти болезни включают в себя болезни Альцгеймера, Паркинсона и Гентингтона, а также боковой амиотрофический склероз, лобно-височные деменции, хроническую травматическую энцефалопатию и диабет второго типа. Различные белки организма-хозяина образуют скопления при этих и многих других заболеваниях, но, как и прионы, такие скопления обычно растут за счет включения в затравку нормальных растворимых молекул белка. Таким образом, потенциал для прионоподобного распространения белков существует на молекулярном уровне. Также растет количество доказательств того, что множество различных связанных с болезнями белковых отложений может расти и распространяться так же, как и прионы, вызывая патологии после инокуляции в локализованные участки у подопытных животных.
Результаты этих исследований поднимают насущные вопросы о том, могут ли многочисленные заболевания, основанные на повторном фолдинге белка, – а они зачастую гораздо более распространены, чем прионные заболевания, основанные на PrP, – быть переданы людям или животным в реальных условиях. Болезнь Крейтцфельдта – Якоба передается между людьми через трансплантацию тканей, инъекции гормонов, полученных от трупов, переливание крови и зараженные медицинские инструменты. Вторым фактором в таких ятрогенных передачах является тот факт, что прионы часто не полностью инактивируются стандартными процедурами клинической дезинфекции.
Еще предстоит установить, могут ли быть определены другие типы потенциально прионоподобных, ассоциированных с болезнями агрегатов белка, которые также могут быть устойчивы к инактивации и при этом способны инициировать или ускорять патогенные процессы у людей. Я не знаю никаких эпидемиологических указаний на то, что это так, но дальнейшее тщательное изучение этого вопроса кажется оправданным.
Об авторе:
Byron W. Caughey – Ph.D., Chief of the TSE/Prion Biochemistry Section of the Laboratory of Persistent Viral Diseases, National Institute of Allergy and Infectious Diseases.
Портал «Вечная молодость» http://vechnayamolodost.ru
Прион – не болезнь и не вирус, а гораздо опаснее
Болезни, вызванные прионами считаются самыми опасными, от них до сих пор нет лекарств. Существо, которое поразил прион обречено.
Прионы не относятся ни к бактериям, ни к вирусам. Ни к грибкам. Это совершено уникальный класс инфекционных агентов, который не содержит в себе нуклеиновых структур. Прионы являются белками с третичной структурой, их размножение происходит путем использования и поедания живых клеток организма. Однако не все организмы могут отвечать требованиям для размножений прионов, а только те у которых присутствует нормальная форма прионного белка. А она есть у человека, крупнорогатого скота и многих других видов.
 Схематичное изображение белка приона
Схематичное изображение белка прионаКакие болезни вызывает прион
У человека это: Крейтцфельдта – Якоба, Куру, синдром Герстмана — Штраусслера и фатальная семейная бессонница. У скота это коровье бешенство. Все эти заболевания объединяет одно – поражение нервной ткани и тканей головного мозга. Из-за деградации тканей имеет схожую клиническую картину с болезнью Альцгеймера.
Основное заражение прионами происходит при поедании зараженной ткани другого организма. Однако есть доказательства, свидетельствующие о том, что прионные агенты содержаться также в навозе и моче. Из-за этого при попадании в водоемы и почву может значительно увеличится скорость распространения таких инфекции.
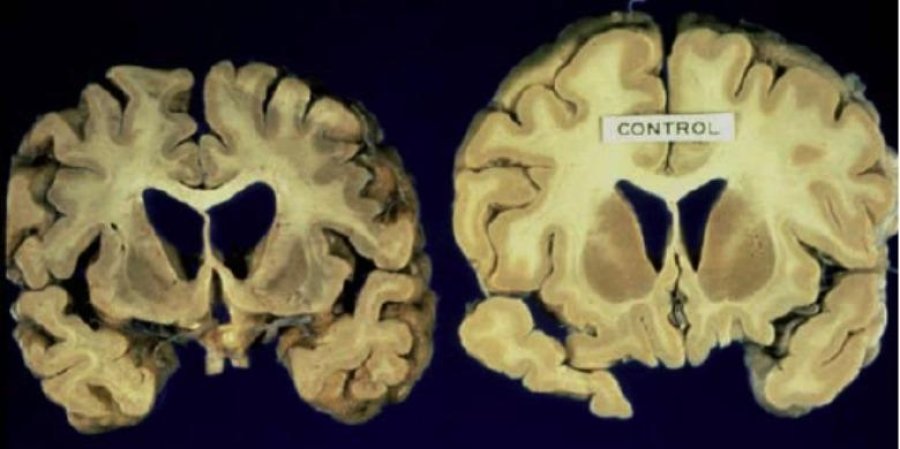 Прион: развитие болезни. Слева пораженный мозг, справа здоровый
Прион: развитие болезни. Слева пораженный мозг, справа здоровыйПрион крайне устойчивая форма белка и лекарств от нее пока не обнаружено. На него не действует радиация (в дозах безопасных для других тканей), а для вирусов он слишком мал. Поэтому при возникновении эпидемии коровьего бешенства люди вынуждены уничтожать все поголовье скота.
В отличии от вирусов и бактерий прион крайне медлителен в размножении. От периода заражения до проявления первых симптомов может пройти до 50 лет, это делает его обнаружение крайне трудоемким.
Поделиться ссылкой:
Прионная болезнь Википедия
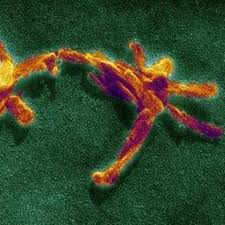
Не следует путать с преонами — гипотетическими элементарными частицами.Прио́ны (англ. prion от protein «белок» + infection «инфекция»; слово было предложено в 1982 году Стенли Прузинером[1]) — особый класс инфекционных патогенов, представленных белками с аномальной третичной структурой, не содержащими нуклеиновые кислоты. Это положение лежит в основе прионной гипотезы[2], однако насчёт состава прионов существуют и другие, маргинальные точки зрения.
Прионы способны увеличивать свою численность, используя функции живых клеток (в этом отношении прионы схожи с вирусами). Прион — это белок с аномальной третичной структурой, способный катализировать конформационное превращение гомологичного ему нормального клеточного белка в себе подобный (прион). Как правило, при переходе белка в прионное состояние его α-спирали превращаются в β-слои. Появившиеся в результате такого перехода прионы могут в свою очередь перестраивать новые молекулы белка; таким образом, запускается цепная реакция, в ходе которой образуется огромное количество неправильно свёрнутых молекул[3]. Прионы — единственные известные инфекционные агенты, размножение которых происходит без участия нуклеиновых кислот.
Все известные прионы вызывают формирование амилоидов — белковых агрегатов, включающих плотно упакованные β-слои. Амилоиды представляют собой фибриллы, растущие на концах, а разлом фибриллы приводит к появлению четырёх растущих концов. Инкубационный период прионного заболевания определяется скоростью экспоненциального роста количества прионов, а она, в свою очередь, зависит от скорости линейного роста и фрагментации агрегатов (фибрилл)[4]. Для размножения приона необходимо исходное наличие нормально уложенного клеточного прионного белка; организмы, у которых отсутствует нормальная форма прионного белка, не страдают прионными заболеваниями.
Прионная форма белка чрезвычайно стабильна и накапливается в поражённой ткани, вызывая её повреждение и, в конечном счёте, отмирание[5]. Стабильность прионной формы означает, что прионы устойчивы к денатурации под действием химических и физических агентов, поэтому уничтожить эти частицы или сдержать их рост тяжело. Прионы существуют в нескольких формах — штаммах, каждый со слегка отличной структурой.
Прионы вызывают заболевания — трансмиссивные губчатые энцефалопатии (ТГЭ) у различных млекопитающих, в том числе
Прионные болезни: диагностика, лечение и виды
Неврология
10.11.2018
 Прионная болезнь — разновидность нейродегенеративных патологий, поражающих головной мозг, особенность состоит в том, что она может развиваться не только в организме человека, но и животного.
Прионная болезнь — разновидность нейродегенеративных патологий, поражающих головной мозг, особенность состоит в том, что она может развиваться не только в организме человека, но и животного.
Возбудителями заболевания являются прионы – молекулы белковой природы, в структуре которых отсутствует нуклеиновая кислота, беспрепятственно проникающие в живой организм и стимулирующие развитие дегенеративно-дистрофических процессов.
Поскольку заболевание нервной системы прогрессирует, его надо своевременно обнаружить и начать лечение, чтобы предотвратить летальный исход.
Что такое прионы?
Существует две формы белковых клеток – нормальная и патологическая. Нормальными белковыми элементами называются клетки, отвечающие за стабилизацию суточных ритмов, укрепление нервных волокон и передачу нервных импульсов. Патологические белки, именуемые прионами, являются результатом аномальной трансформации нормальных клеток.
Прионами называют опасные инфекционные агенты белкового типа, не принадлежащие к группе живых микроорганизмов, в их структуре не содержатся такие молекулы, как РНК и ДНК. Характерная особенность прионов в следующем: несмотря на то, что в строении белковых возбудителей отсутствуют нуклеиновые кислоты, они с легкостью размножаются, попадая в организм и поражая здоровые клетки.
Уязвимыми перед воздействием прионных возбудителей считаются нейроциты, что обуславливает поражение нервной системы: в наименьшей степени поражаются клетки, из которых сформирована лимфатическая ткань. Диагностировать патологию своевременно сложно, период от проникновения инфекции в организм до начала развития болезни составляет не менее года.
Свойства прионных болезней
 Прионы и прионные болезни обладают высокой устойчивостью: такие методы, как длительное кипячение при высокой температуре и воздействие холодом оказываются неэффективными в борьбе с ними.
Прионы и прионные болезни обладают высокой устойчивостью: такие методы, как длительное кипячение при высокой температуре и воздействие холодом оказываются неэффективными в борьбе с ними.
Невозможно уничтожить белковые возбудители также ультрафиолетовым излучением, радиацией и формалиновой обработкой.
Структура инфекционных агентов обуславливает снижение выработки антител, отвечающих за формирование гуморального иммунитета, а также лимфоцитов, нехватка которых провоцирует снижение клеточного иммунитета.
Угнетение иммунной системы предшествует тому, что живой организм перестает бороться с патологическими молекулами и игнорирует, вследствие этого прионы продолжают заражать клетки.
Процесс развития прионной болезни
В первую очередь замещение здоровых белковых клеток происходит в лимфатической и дендритной ткани, селезенке, а также тимусе. Накапливаясь в клеточной структуре, инфекционные агенты начинают передвигаться по организму аксональным методом и посредством нервных стволов проникают в головной мозг.
Поскольку ослабленный зараженный организм не реагирует на внедрение опасных инфекций, заболевание принимает прогрессирующую форму и усугубляется патологическими процессами:
- формирование полостей в клеточной паренхиме головного мозга;
- уничтожение нейронов, в результате происходит расстройство работы нервной системы;
- появление амилоидных бляшек, которые представляют собой скопление белковых прионов;
- замещение отмерших нервных волокон соединительной тканью.
При отсутствии лечения дегенеративно-дистрофические отклонения переходят в хроническую стадию, что стимулирует атрофию мозговых клеток, повышает вероятность летального исхода.
Как можно заразиться?
Различают три основных пути заражения прионными белками:
- Трансмиссивный способ основывается на том, что вирусы передаются от инфицированного носителя неинфицированному организму в результате потребления зараженного мяса и молока, а также использования зараженного биоматериала – крови или роговицы.
- Наследственный способ предполагает, что прионная болезнь начинает развиваться от аномальной мутации 20-й хромосомы, которая отвечают за синтез белка.
- Спорадический способ заражения характеризуется замещением здоровых белковых тел аномальными клетками.
Вне зависимости от того, какую природу имеет патология, первичную или вторичную, она может передаваться здоровым организмам трансмиссивным путем. Это объясняет тот факт, что игнорировать заболевание нельзя.
Классификация болезней
К прионным болезням человека относят заболевание Крейтцфельда-Якоба, патология куру, болезнь Альперса, семейную фатальную инсомнию, болезнь Герстманна. Каждая из патологий имеет особенности течения, что обуславливает наличие отличий в методике их лечения.
Заболевание Крейтцфельда-Якоба
Данная болезнь принадлежит к группе заболеваний мозга дегенеративного характера и характеризуется прогрессирующим некрозом нейронов.
 На ранних стадиях развития патологии у больного наблюдаются изменения личностных качеств: он становится замкнутым и пребывает в депрессивном состоянии. Прионное заболевание провоцирует расстройство речевой, слуховой и функции зрения, снижает интеллект и способствует развитию галлюцинаций.
На ранних стадиях развития патологии у больного наблюдаются изменения личностных качеств: он становится замкнутым и пребывает в депрессивном состоянии. Прионное заболевание провоцирует расстройство речевой, слуховой и функции зрения, снижает интеллект и способствует развитию галлюцинаций.
В зависимости от особенностей развития, различают формы болезни Крейтцфельда-Якоба:
- спорадическая форма считается распространенной и диагностируется преимущественно у людей преклонного возраста. Проявление первых симптомов происходит в возрасте 55 лет: пациент жалуется на головную боль, бессонницу, отсутствие аппетита и общую слабость. По мере развития болезни у больного начинают проявляться неврологические расстройства: повышается тонус мышц, нарушается речевой аппарат, возникают судороги;
- семейная форма образуется как результат аномальной мутации 20-й хромосомы. Несмотря на то, что симптоматика этой формы схожа с клинической картиной, которая проявляется при спорадическом типе, она проявляется на 10 лет раньше, когда больной пребывает в зрелом возрасте;
- ятрогенная форма предполагает заражение живого организма хирургическим путем. Длительность инкубационного периода определяется на основании того, как аномальные клетки попали в клеточную структуру. Наиболее быстро прионное заболевание проявляется, если заражение тканей произошло в результате оперирования мозговых тканей, наименее быстро – пересадки роговицы.
Помимо трех форм патологии, существует атипичный вид, когда заражение аномальным белком происходит в процессе потребления человеком инфицированных мясных продуктов.
Болезнь куру
Уязвимыми перед развитием заболевания были аборигены, образ жизни которых предполагал проведение ритуалов каннибализма. После завершения инкубационного периода людоеды отмечали проявление таких симптомов, как непроизвольное дрожание конечностей, мышечная слабость, невнятная речь, неконтролируемый смех и затрудненное глотание.
В наши дни болезнь куру не диагностируется, что обусловлено отменой традиций каннибализма, запретом проведения соответствующих ритуалов.
Болезнь Альперса
Заболевание имеет наследственную природу и проявляется у детей до 18 лет. Основным осложнением патологии считается поражение нервной системы, вследствие чего у ребенка ухудшается зрение, развиваются эпилептические припадки, которые периодически обостряются.
Помимо неврологического расстройства, болезнь Альперса поражает печень больного, что повышает риск развития хронической формы гепатита или цирроза. При отсутствии лечения на фоне сопутствующих заболеваний образуется печеночная недостаточность: как результат организм ребенка страдает от интоксикации.
Семейная фатальная инсомния
Эта прионная болезнь передается аутосомно-доминантным путем, имеет стадии развития:
- Первая стадия характеризуется проявлением у больного фобий страхов. Её длительность не превышает 4 месяцев.
- Вторая стадия характеризуется присутствием беспричинной тревоги и частых галлюцинаций. В среднем продолжительность данной стадии составляет 5 месяцев.
- Третья стадия длится не более месяца и характеризуется развитием психических отклонений, обусловленных хронической бессонницей.
- Четвертая стадия диагностируется, если полное отсутствие сна усугубляется деменцией. Её продолжительность составляет полгода, после чего пациент умирает в результате сильного истощения.
По сравнению с другими видами прионных заболеваний, семейная фатальная инсомния диагностируется очень редко.
Болезнь Герстманна
Заболевание наследственного характера проявляется в возрасте 40 лет и характеризуется медленным развитием: с момента заражения организма прионами до гибели больного проходит не менее 5 лет. Клиническая картина состоит из таких симптомов, как ослабление зрения и слуха, затрудненное глотание, повышение мышечного тонуса, а также ограниченная двигательная активность.
Как диагностируют и лечат патологию
При наличии признаков развития прионного заболевания пациенту нужно обратиться к врачу. Во время осмотра доктор изучит анамнез больного на предмет проведения пересадки органов и посредством устного опроса уточнит сферу деятельности и особенности образа жизни.
 Врач направляет пациента на сдачу общего анализа крови, также забор спинномозгового вещества. Чтобы исследование нервных тканей и головного мозга было результативным, лабораторная диагностика дополняется инструментальными методами – компьютерной и магнитно-резонансной томографией.
Врач направляет пациента на сдачу общего анализа крови, также забор спинномозгового вещества. Чтобы исследование нервных тканей и головного мозга было результативным, лабораторная диагностика дополняется инструментальными методами – компьютерной и магнитно-резонансной томографией.
Поскольку прионные заболевания – патологии, сопровождающиеся необратимыми дегенеративными изменениями, вылечить их невозможно. Основываясь на результатах диагностики, врач составит схему терапии, цель которой улучшить состояние пациента посредством приема успокоительных и противосудорожных препаратов.
Профилактические меры
Избежать инфицирования прионным заболеванием трансмиссионным путем возможно. С это целью нужно соблюдать простые правила:
- Потреблять качественное мясо и молочные изделия: отказаться от покупки продуктов животного происхождения, поставляемые регионами, в которых зарегистрированы случаи заражения прионами.
- С осторожностью принимать лекарственные препараты, в состав которых входит человеческая или животная кровь: по возможности рекомендуется заменять их синтетическими аналогами.
Предотвратить заражение прионными молекулами, которые являются результатом генетической аномалии, невозможно. Данный факт обусловлен тем, что механизм развития такой формы имеет наследственный характер.
прионных болезней | Johns Hopkins Medicine
Что такое прионные болезни?
Прионные болезни включают несколько состояний. Прион — это тип белка, который может вызвать аномальное сворачивание нормальных белков в мозге. Прионные болезни могут поражать как людей, так и животных и иногда передаются людям через инфицированные мясные продукты. Наиболее распространенной формой прионной болезни, поражающей людей, является болезнь Крейтцфельдта-Якоба (CJD).
Прионные болезни встречаются редко. Ежегодно в США регистрируется около 300 случаев заболевания.С.
К видам прионных болезней относятся:
- CJD. Человек может унаследовать это состояние, и в этом случае оно называется семейным CJD. С другой стороны, спорадическая CJD развивается внезапно без каких-либо известных факторов риска. Большинство случаев CJD носят спорадический характер и, как правило, поражают людей в возрасте около 60 лет. Приобретенная CJD возникает в результате контакта с инфицированной тканью во время медицинской процедуры, такой как трансплантация роговицы. Симптомы CJD (см. Ниже) быстро приводят к тяжелой инвалидности и смерти.В большинстве случаев смерть наступает в течение года.
- Вариант CJD. Это инфекционный тип болезни, относящейся к «коровьему бешенству». Употребление в пищу больного мяса может вызвать заболевание у человека. Мясо может вызвать аномальное развитие нормального человеческого прионного белка. Этот тип заболевания обычно поражает людей более молодого возраста.
- Вариативная протеаза — Чувствительная прионопатия (VPSPr). Это тоже крайне редко, похоже на CJD, но белок менее чувствителен к пищеварению.Это чаще встречается у людей в возрасте около 70 лет, у которых есть семейная история деменции.
- Gerstmann- Болезнь Штройсслера-Шейнкера (GSS). Чрезвычайно редко, но встречается в более раннем возрасте, обычно около 40 лет.
- Куру. Заболевание обнаружено в Новой Гвинее. Это вызвано употреблением в пищу тканей человеческого мозга, зараженных инфекционными прионами. Из-за повышения осведомленности о болезни и о том, как она передается, куру сейчас встречается редко.
- Смертельная бессонница (FI). Редкое наследственное заболевание, вызывающее нарушение сна. Также существует спорадическая форма заболевания, не передающаяся по наследству.
Что вызывает прионную болезнь?
Прионные заболевания возникают, когда нормальный прионный белок, обнаруженный на поверхности многих клеток, становится аномальным и скапливается в головном мозге, вызывая повреждение мозга. Это ненормальное накопление белка в головном мозге может вызвать нарушение памяти, изменения личности и трудности с движением.Специалисты до сих пор мало знают о прионных заболеваниях, но, к сожалению, эти нарушения обычно приводят к летальному исходу.
Кто подвержен риску прионных заболеваний?
Факторы риска прионной болезни включают:- Семейный анамнез прионной болезни
- Употребление в пищу мяса, зараженного «коровьим бешенством»
- Инфекция в результате заражения роговицы или зараженного медицинского оборудования
Каковы симптомы прионных болезней?
Симптомы прионных болезней включают:
- Быстро развивающаяся деменция
- Затруднение при ходьбе и изменения походки
- Галлюцинации
- Жесткость мышц
- Путаница
- Усталость
- Затруднения при разговоре
Как диагностируются прионные заболевания?
Прионные заболевания подтверждаются взятием образца ткани мозга во время биопсии или после смерти.Однако перед этим медицинские работники могут провести ряд тестов, чтобы помочь диагностировать прионные заболевания, такие как CJD, или исключить другие заболевания с аналогичными симптомами. Прионные заболевания следует рассматривать у всех людей с быстро прогрессирующей деменцией.
Испытания включают:
- МРТ (магнитно-резонансная томография) головного мозга
- Образцы жидкости из спинного мозга (спинномозговая пункция, также называемая люмбальной пункцией)
- Электроэнцефалограмма, анализирующая мозговые волны; этот безболезненный тест требует наложения электродов на кожу головы
- Анализы крови
- Неврологический и визуальный осмотр для проверки повреждения нервов и потери зрения
Как лечат прионные заболевания?
Прионные болезни нельзя вылечить, но некоторые лекарства могут замедлить их прогрессирование.Медицинское управление направлено на обеспечение максимальной безопасности и комфорта людей с этими заболеваниями, несмотря на прогрессирующие и изнурительные симптомы.
Можно ли предотвратить прионные заболевания?
Правильная очистка и стерилизация медицинского оборудования может предотвратить распространение болезни. Если у вас есть или может быть CJD, не жертвуйте органы или ткани, включая ткань роговицы.
Новые правила содержания и кормления коров могут помочь предотвратить распространение прионных заболеваний.
Живущие с прионными заболеваниями
По мере прогрессирования прионных заболеваний людям с этими заболеваниями обычно требуется помощь в уходе за собой. В некоторых случаях они могут оставаться в своих домах, но в конечном итоге им может потребоваться переехать в учреждение по уходу.
Основные сведения о прионных заболеваниях
- Прионные болезни очень редки.
- Симптомы могут быстро прогрессировать, что требует повседневной помощи.
- Прионные болезни всегда смертельны.
Следующие шаги
Советы, которые помогут вам получить максимальную пользу от посещения врача:
- Знайте причину вашего визита и то, что вы хотите.
- Перед визитом запишите вопросы, на которые хотите получить ответы.
- Возьмите с собой кого-нибудь, кто поможет вам задать вопросы и запомнить, что вам говорит поставщик.
- Во время посещения запишите название нового диагноза и любые новые лекарства, методы лечения или тесты.Также запишите все новые инструкции, которые дает вам ваш провайдер.
- Узнайте, почему прописано новое лекарство или лечение и как они вам помогут. Также знайте, каковы побочные эффекты.
- Спросите, можно ли вылечить ваше состояние другими способами.
- Знайте, почему рекомендуется тест или процедура и что могут означать результаты.
- Знайте, чего ожидать, если вы не примете лекарство, не пройдете анализ или процедуру.
- Если вам назначена повторная встреча, запишите дату, время и цель этого визита.
- Знайте, как вы можете связаться с вашим провайдером, если у вас есть вопросы.
Прионная болезнь — Genetics Home Reference
От 10 до 15 процентов всех случаев прионной болезни вызваны мутациями в гене PRNP. Поскольку они могут передаваться по наследству, эти формы прионной болезни классифицируются как семейные. Семейные прионные заболевания, признаки и симптомы которых перекрывают друг друга, включают семейную болезнь Крейтцфельдта-Якоба (CJD), синдром Герстманна-Штройсслера-Шейнкера (GSS) и фатальную семейную бессонницу (FFI).
Ген PRNP предоставляет инструкции по созданию белка, называемого прионным белком (PrP).Хотя точная функция этого белка неизвестна, исследователи предложили роль в нескольких важных процессах. К ним относятся транспорт меди в клетки, защита клеток мозга (нейронов) от повреждений (нейрозащита) и связь между нейронами. При семейных формах прионного заболевания мутации гена PRNP приводят к образованию PrP Sc из одной копии гена. В процессе, который до конца не изучен, PrP Sc может присоединяться (связываться) с нормальным белком (PrP C ) и способствовать его превращению в PrP Sc .Аномальный белок накапливается в мозгу, образуя сгустки, которые повреждают или разрушают нейроны. Утрата этих клеток приводит к образованию микроскопических губчатых отверстий (вакуолей) в головном мозге, что приводит к появлению признаков и симптомов прионной болезни.
Остальные 85–90 процентов случаев прионной болезни классифицируются как спорадические или приобретенные. Люди со спорадической прионной болезнью не имеют семейного анамнеза болезни и не имеют выявленной мутации в гене PRNP. Спорадическое заболевание возникает, когда PrP C спонтанно и по неизвестным причинам превращается в PrP Sc .Спорадические формы прионной болезни включают спорадическую болезнь Крейтцфельда-Якоба (sCJD), спорадическую фатальную бессонницу (sFI) и прионопатию, чувствительную к различным протеазам (VPSPr).
Приобретенная прионная болезнь возникает в результате воздействия PrP Sc из внешнего источника. Например, вариант болезни Крейтцфельдта-Якоба (vCJD) представляет собой тип приобретенного прионного заболевания у людей, который возникает в результате употребления в пищу продуктов из говядины, содержащих PrP Sc , от крупного рогатого скота с прионной болезнью. У коров эта форма заболевания известна как губчатая энцефалопатия крупного рогатого скота (BSE) или, чаще, «коровье бешенство».Другим примером приобретенного прионного заболевания человека является куру, которое было выявлено в популяции Саут-Форе в Папуа-Новой Гвинее. Заболевание передавалось, когда люди ели пораженные человеческие ткани во время каннибалистических похоронных ритуалов.
Редко прионная болезнь может передаваться через случайное воздействие тканей, загрязненных PrP Sc , во время медицинской процедуры.Этот тип прионной болезни, составляющий от 1 до 2 процентов всех случаев, классифицируется как ятрогенный.
.Прионная болезнь — Simple English Wikipedia, бесплатная энциклопедия
Прионная болезнь (также называемая трансмиссивной губчатой энцефалопатией ) — это болезнь, вызываемая прионами. Прионы представляют собой структурно измененные версии небольших белков, которые обычно экспрессируются в клетках. В отличие от заболеваний, которые вызваны мутацией гена, приводящей к экспрессии мутантного белка, прионы способны реплицироваться и передавать заболевания посредством физического контакта с нормальными белками, что приводит к структурному изменению из нормального состояния в состояние приона.
В отличие от бактерий, прионы не считаются живыми, потому что у них нет собственного метаболизма, они не обладают генами и не могут естественным образом воспроизводиться вне клетки-хозяина. Прионные заболевания очень редки, и для большинства из них нет лечения.
Практически все известные прионные заболевания являются неврологическими. Есть два общих признака, которые наблюдаются при типичных прионных заболеваниях:
- Атаксия или нарушение равновесия — это когда пациент не может хорошо стоять или ходить, потому что не может поддерживать свое равновесие.Обычно это происходит из-за заболевания мозжечка.
- Деменция или потеря психики — это прогрессирующая потеря когнитивных функций.
Известно несколько видов прионных болезней. Наиболее важные из них:
- Болезнь Крейтцфельдта-Якоба — Человек может заразиться, если съел говядину, инфицированную BSE, получив переливание крови от больного, инъекцию гормона роста человека, выделенного из тел с болезнью, или инфицированные хирургическим путем. инструменты.Пациенты страдают атаксией, слабоумием и обычно умирают через год.
- Болезнь Куру — Это заболевание встречается у людей, живущих в Новой Гвинее, которые поедают мозги мертвых людей. Пациенты страдают атаксией, деменцией и неспособностью двигать глазами. Обычно они умирают в течение двух лет.
- Синдром Герстмана-Штрауслера-Шейнкера — это очень редкий синдром, который проявляется атаксией и деменцией. Пациенты обычно умирают через год.
Прионные болезни очень редки.Болезнь Крейтцфельдта-Якоба, которая составляет около 85% случаев, составляет от 1 до 1,5 случаев на миллион человек в год.
.ВОЗ | Прионные болезни
Губчатая энцефалопатия крупного рогатого скота (ГЭКРС) является недавно зарегистрированной трансмиссивной губчатой энцефалопатией (ГЭ) крупного рогатого скота. Заболевание человека «вариант болезни Крейтцфельда-Якоба» (vCJD) считается зоонозным заболеванием, вызываемым агентом BSE. С октября 1996 г. по ноябрь 2004 г. было зарегистрировано 152 случая (вероятных) вБКЯ в Великобритании, восемь во Франции, два в Ирландии и по одному в Канаде, Италии и Соединенных Штатах Америки.В настоящее время имеется недостаточно информации, чтобы сделать обоснованный прогноз относительно будущего числа случаев vCJD.
Путь передачи vCJD еще полностью не доказан, но общепризнано, что он передается через пищу, загрязненную возбудителем BSE. В период с 1986 по октябрь 2004 года в Соединенном Королевстве было подтверждено более 180 000 случаев BSE у крупного рогатого скота. Болезнь также была обнаружена в нескольких других странах. Передача BSE происходит, когда крупный рогатый скот ест мясокостную муку, зараженную возбудителем.
Были определены и введены в действие меры по предотвращению воздействия на человека в Великобритании и других странах. Эти меры включают запрет на использование мясокостной муки в кормах для животных, тестирование убитых животных, систематическое удаление «материалов высокого риска» из туш и уничтожение подозрительных и подтвержденных случаев крупного рогатого скота, а также контроль животных, потенциально подвергшихся заражению в то же время. время.
Скрепи, другое животное, TSE, является эндемическим для некоторых стад овец и коз в Европе, Азии и Северной Америке (возможно, в других местах), за исключением небольшого числа стран.В течение столетий сожительства людей и животных никогда не было доказанной опасности заражения овец для людей. Недавно TSE других животных были обнаружены у лосей и оленей (хроническая истощающая болезнь, CWD), норок (трансмиссивная норковая энцефалопатия, TME) и кошачьих (Feline Spongiform Encephalopathy, FSE), но до сих пор ни один случай заболевания человека не был связан с этими заболеваниями животных. .
TSE человека встречаются в спорадических, семейных и приобретенных формах. Наиболее распространенная форма, спорадическая болезнь Крейтцфельдта-Якоба (БКЯ), имеет во всем мире уровень смертности около 1 случая на миллион человек в год и является причиной около 85% всех случаев БКЯ во всем мире.Причина спорадической CJD остается неизвестной. Семейная болезнь Крейтцфельдта-Якоба (fCJD) передается по наследству в результате генетических мутаций и составляет 10-15% случаев во всем мире. Остальные случаи — ятрогенная и вариантная болезнь Крейтцфельдта-Якоба.
Куру был известен как человеческий TSE в ограниченном районе Папуа-Новой Гвинеи с распространенностью до 2% в некоторых племенах. Сейчас встречается редко. Нет свидетельств, указывающих на распространение куру каким-либо другим механизмом, кроме ритуального каннибализма.
.