Прионная болезнь что это такое: Прионные болезни
Авторизация
Размер:
AAA
Цвет: C C C
Изображения Вкл. Выкл.
Обычная версия сайта
Сведения об образовательной организации Контакты Старая версия сайта Версия для слабовидящих Версия для слабовидящих
Южно-Уральский государственный медицинский университет
- Университет
- События
- Новости
- ЮУГМУ сегодня
- Историческая справка
- Руководство
- Выборы ректора
- Лицензия, аккредитация и сертификаты
- Организационная структура
- Противодействие коррупции
- Первичная профсоюзная организация ЮУГМУ Профсоюза работников здравоохранения РФ
- Абитуриенту
- Новости для абитуриентов
- Центр довузовской подготовки
- Поступающим на специалитет
- Поступающим в ординатуру
- Поступающим в аспирантуру
- Поступающим в медицинский колледж
- Документы на право ведения образовательной деятельности
- Положения о приемной, экзаменационной и апелляционной комиссиях
- Информация об общежитиях
- Часто задаваемые вопросы
- Результаты приема студентов
- Информация для инвалидов
- Обучающемуся
- Факультеты
- Кафедры
- Медицинский колледж
- Ординатура
- Аспирантура
- Научная библиотека
- Образовательный портал
- Расписания
- Этический кодекс студентов медицинских вузов
- Совет студентов Минздрава России
- О допуске студентов к работе в медицинских организациях
- Иностранным обучающимся
- Медицинское обслуживание
- Информация об общежитиях
- Стипендиальное обеспечение
- Порядок перехода обучающихся с платного на бесплатное обучение
- Часто задаваемые вопросы
- Анкетирование
- Студенческие отряды
- Противодействие терроризму и экстремизму
- Специалисту
- Институт дополнительного профессионального образования
- Аккредитация специалистов
- Пациенту
- Клиника ФГБОУ ВО ЮУГМУ Минздрава России
- Профилактика новой коронавирусной инфекции, гриппа, ОРВИ, вакцинация
- Здоровый образ жизни
- Нет наркотикам!
- Научная работа
- Управление по научной и инновационной работе
- Экспериментально-биологическая клиника (виварий)
- Отдел международных связей
- Центральная научно-исследовательская лаборатория
- НОЦ «Проблемы фундаментальной медицины»
- НОЦ «Клиническая фармакология»
- Конференции и другие мероприятия
- Диссертационные советы
- Журнал «Непрерывное медицинское образование и наука»
- Студенческое научное общество
- Совет молодых ученых и специалистов
Прионы: смертоносные молекулы-зомби
Роман Фишман
«Популярная механика» №11, 2015
«Самое страшное в них то, что они действуют не как хищники, а как вирусы. Хищники по природе своей разумны и не уничтожают всех жертв поголовно… А они — просто размножаются, заражают и пожирают. Все остальное им совершенно безразлично», — так объясняет свой ужас перед зомби автор бестселлера «Мировая война Z» Макс Брукс. Но то же можно сказать и о прионах — настоящих смертоносных молекулах-зомби.
Хищники по природе своей разумны и не уничтожают всех жертв поголовно… А они — просто размножаются, заражают и пожирают. Все остальное им совершенно безразлично», — так объясняет свой ужас перед зомби автор бестселлера «Мировая война Z» Макс Брукс. Но то же можно сказать и о прионах — настоящих смертоносных молекулах-зомби.
Сравнение с ходячими мертвецами напрашивается. Как и они, прионы в буквальном смысле слова разрушают мозг, превращая человека сперва в овощ, а затем — в труп. Как зомби из людей, так и они появляются из самых обычных белков. Их крайне сложно уничтожить, зато сами они смертельны в ста процентах случаев. Впрочем, трудно было бы ожидать иного от инфекционных агентов, которых вообще нельзя назвать живыми. Несколько столетий они ускользали от ученых — и даже потом в их существование поверили далеко не сразу. А теперь в них иногда видят даже источник жизни.
Овцы и людоеды
Первое «нашествие зомби» отмечено на рубеже XVII и XVIII веков, когда в Англии вовсю громыхала промышленная революция. Среди огромных стад, снабжавших шерстью быстрорастущие текстильные предприятия, то и дело стали попадаться «паршивые овцы». Напасть развивалась медленно, но неотвратимо: животные мучительно, до крови чесались об ограду, затем нарушалась координация движений, чаще и чаще случались судороги — через неделю-месяц все заканчивалось смертью.
Среди огромных стад, снабжавших шерстью быстрорастущие текстильные предприятия, то и дело стали попадаться «паршивые овцы». Напасть развивалась медленно, но неотвратимо: животные мучительно, до крови чесались об ограду, затем нарушалась координация движений, чаще и чаще случались судороги — через неделю-месяц все заканчивалось смертью.
Прошли столетия, Пастер описал бактериальные инфекции, а после работ Ивановского и Бейеринка появились представления и о вирусных заболеваниях. Но «почесуха овец», или скрейпи, оставалась загадкой. Все говорило о поражении мозга, ткани которого болезнь превращала в нечто, похожее на губку, изъеденную неровными порами. Но на вопрос о ее возбудителе специалистам оставалось лишь разводить руками: ни бактерий, ни вирусов найдено не было. Зато нашлись у скрейпи последователи.
В одном и том же 1920 году, но независимо друг от друга, Ганс Крейтцфельдт и Альфонс Якоб описали неизлечимое и неумолимое поражение нервной системы человека. Возникая по неизвестной причине, обычно в пожилом возрасте, болезнь часто начиналась проблемами со сном и ослаблением когнитивных функций, развивалась потерей координации движений и деменцией, а несколько лет спустя венчалась параличом и функциональными нарушениями, окончательно несовместимыми с жизнью. Ткани мозга снова напоминали губку — и снова никаких следов возбудителя.
Ткани мозга снова напоминали губку — и снова никаких следов возбудителя.
Редкое заболевание могло изучаться еще долго и неторопливо, не создавая особенного ажиотажа, если бы у него не обнаружился странный родственник в противоположной части света. В начале 1954 года чиновники Новой Гвинеи, тогда еще бывшей частью Австралии, составили отчет, в котором сообщали о вспышке диковинной местной болезни куру. «Первым признаком надвигающейся смерти становится слабоумие, за которым следует общая слабость вплоть до неспособности самостоятельно стоять на ногах, — говорилось в сообщении. — На следующем этапе жертва лежит, не в силах даже принять помощь, пока, наконец, не наступит гибель». Описанием болезни занялись медики Винсент Зигас и Карлтон Гайдушек, выяснившие, что поражает она лишь одно из новогвинейских племен, форе, выделяющееся среди соседей своим пристрастием к каннибализму. Вряд ли можно считать эту традицию какой-то особенно жестокой: существование в условиях серьезного дефицита жизненных ресурсов порождает и не такие обычаи. Так что после смерти аборигена форе его родственники поедали останки, причем по традиции мясо доставалось мужчинам, а женщины и дети довольствовались остальным, в том числе и мозгом. Как правило, заболевали именно они, причем мозг поражался в первую очередь: губчатая масса переродившейся нервной ткани была все той же, что и при скрейпи овец, и при болезни Крейтцфельдта — Якоба. Так что вскоре врачи решили, что имеют дело с медленной вирусной инфекцией, которая поражает нервную ткань и передается алиментарным путем (как «зомбированность»), через поедание больного мозга. Только вот выделить вирус снова никак не удавалось.
Так что после смерти аборигена форе его родственники поедали останки, причем по традиции мясо доставалось мужчинам, а женщины и дети довольствовались остальным, в том числе и мозгом. Как правило, заболевали именно они, причем мозг поражался в первую очередь: губчатая масса переродившейся нервной ткани была все той же, что и при скрейпи овец, и при болезни Крейтцфельдта — Якоба. Так что вскоре врачи решили, что имеют дело с медленной вирусной инфекцией, которая поражает нервную ткань и передается алиментарным путем (как «зомбированность»), через поедание больного мозга. Только вот выделить вирус снова никак не удавалось.
Коровы и радиация
Сходство болезней (необычно долгий инкубационный период и неизбежность летального исхода, заразность и, конечно, характерные перерождения нервной ткани мозга) позволило предположить у них и общую причину и объединить в группу трансмиссивных губчатых энцефалопатий.
Уже в 1960-х британские ученые предположили, что источником их может оказаться белок: таинственный инфекционный агент не инактивировался смертельной даже для вирусов дозой излучения. Это можно было объяснить крошечным размером его частиц, намного меньших, чем даже вирусы. Гипотеза показалась не лишенной смысла, хотя в корне противоречила «основной догме молекулярной биологии», которая постулировала однонаправленный поток информации, свойственный всему живому: от ДНК через РНК к белку. Возможно ли выбросить из этой короткой цепочки целых два звена — и сохранить многие свойства живого?
Это можно было объяснить крошечным размером его частиц, намного меньших, чем даже вирусы. Гипотеза показалась не лишенной смысла, хотя в корне противоречила «основной догме молекулярной биологии», которая постулировала однонаправленный поток информации, свойственный всему живому: от ДНК через РНК к белку. Возможно ли выбросить из этой короткой цепочки целых два звена — и сохранить многие свойства живого?
В 1970-е поисками возбудителя занялся калифорнийский невролог Стэнли Прузинер. Работа шла небыстро: используя биологические жидкости больных скрейпи овец, ученые подтвердили, что болезнь неумолима и развивается у всех до единой зараженных мышей, хотя инкубационный период затягивается на полгода, а то и дольше. Зато результаты многолетних экспериментов оказались сенсационными: снова и снова, выделяя прежде неуловимый инфекционный агент, ученые убеждались, что это «голый» белок.
Принять такое удалось не сразу. Шутка ли: патоген, не имеющий даже намека на ДНК, а между тем — инфекционный, размножающийся и, как покажут дальнейшие исследования, даже мутирующий. Признание случилось во многом по пословице: «Не было бы счастья, да несчастье помогло». В конце 1980-х в Великобритании разразилась эпидемия губчатой энцефалопатии крупного рогатого скота — «коровьего бешенства». Болезнь распространилась так широко, что за следующие годы ее обнаружили почти у 200 000 голов, было зафиксировано несколько сотен случаев передачи ее людям. И это, определенно, был белок: коровам он мог передаться от овец или появиться в сообществе случайно, а затем распространиться через кормовые добавки, произведенные из костной муки забитых животных. В результате в 1990-х прионы, как назвал смертоносные белки Прузинер, скрестивший слова «протеин» и «инфекция», получили почти всеобщее признание, а в 1997 году сам ученый удостоился Нобелевской премии. Вызывавший губчатые энцефалопатии «прионный белок скрейпи» PrP
Признание случилось во многом по пословице: «Не было бы счастья, да несчастье помогло». В конце 1980-х в Великобритании разразилась эпидемия губчатой энцефалопатии крупного рогатого скота — «коровьего бешенства». Болезнь распространилась так широко, что за следующие годы ее обнаружили почти у 200 000 голов, было зафиксировано несколько сотен случаев передачи ее людям. И это, определенно, был белок: коровам он мог передаться от овец или появиться в сообществе случайно, а затем распространиться через кормовые добавки, произведенные из костной муки забитых животных. В результате в 1990-х прионы, как назвал смертоносные белки Прузинер, скрестивший слова «протеин» и «инфекция», получили почти всеобщее признание, а в 1997 году сам ученый удостоился Нобелевской премии. Вызывавший губчатые энцефалопатии «прионный белок скрейпи» PrP Он встречается на мембранах клеток во многих частях тела и тканях здоровых людей и животных, хотя больше всего — в клетках нервной системы, как в самих нейронах, так и в поддерживающих их клетках нейроглии. Судя по всему, он исключительно важен, но для чего?
Он встречается на мембранах клеток во многих частях тела и тканях здоровых людей и животных, хотя больше всего — в клетках нервной системы, как в самих нейронах, так и в поддерживающих их клетках нейроглии. Судя по всему, он исключительно важен, но для чего?
Явление зомби
Было показано, что PrPС обладает высоким сродством с ионами меди, что может указывать на его возможную роль в удалении из клетки токсичных для нее тяжелых металлов. С другой стороны, максимальная концентрация PrPС наблюдается у контактов между нейронами некоторых областей мозга. Клеточные мембраны здесь буквально усеяны этим белком, что, возможно, говорит о том, что он играет какую-то роль в формировании или стабилизации синаптических контактов. Другая группа гипотез говорит о том, что PrPС необходим для удаления «загрязнений» с поверхности клеток. Связывая их, белок меняет форму, и, когда на мембране нейрона накопится достаточно много таких измененных белков, в нем запускается механизм уничтожения, а клетка гибнет.
«Существует, наверное, 20 или 30 гипотез о том, какую именно задачу может выполнять нормальная клеточная форма прионного белка. Но какой-то определенной, четкой функции у него не найдено, поэтому и возникают дебаты, — рассказал нам профессор Медицинской школы Мэриленда Илья Баскаков, не один год посвятивший изучению прионов и прионных болезней. — В последние годы активно обсуждается возможная роль PrPС в процессе развития нервной системы. Он может быть необходим для того, чтобы из стволовых клеток созревали новые нейроны — эксперименты показали, что, если у стволовой клетки выключен кодирующий этот белок ген PRNP, она не может превратиться в нервную».
Функции здорового белка остаются неизвестными, но они явно очень важны: ген PRNP характеризуется высокой консервативностью и мало отличается у людей и других млекопитающих. Теоретически это и позволяет приону передаваться между любыми видами животных, имеющими PrPС, — недаром губчатые энцефалопатии зафиксированы не только у людей, овец и коров, но и у кошек, норок, антилоп, оленей и даже страусов.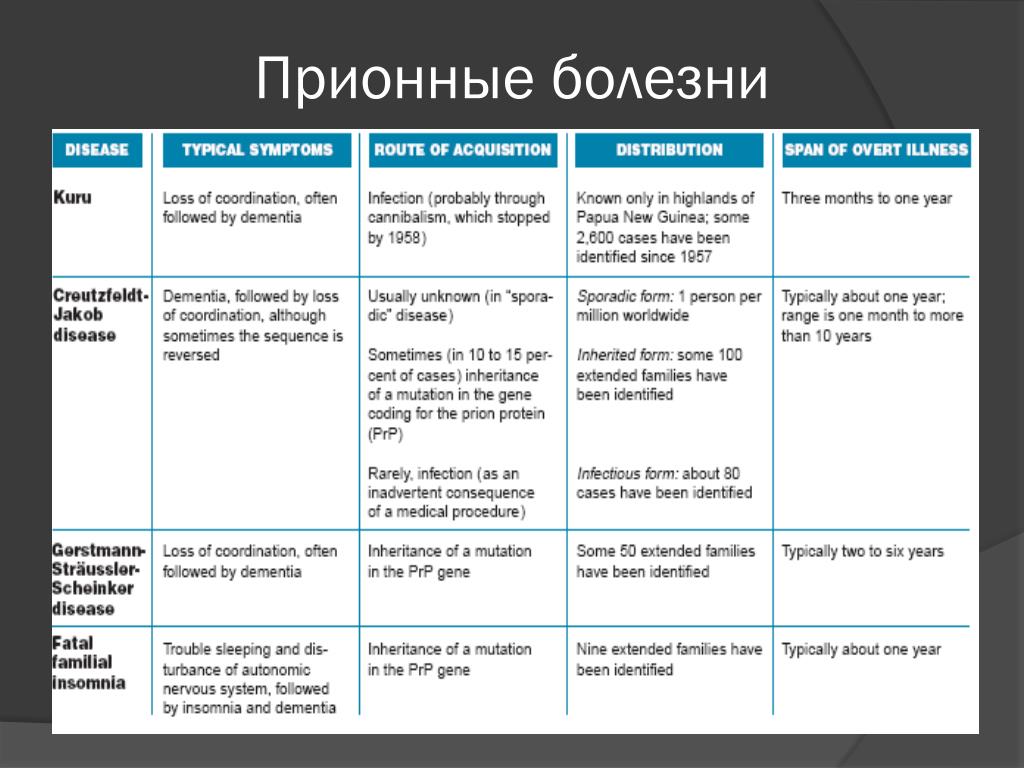 Предполагается, что первый белок-«зомби» в популяции может появиться случайно, в результате неправильно сложившейся пространственной формы здорового белка PrPС. Эта ошибка, на первый взгляд незначительная, меняет все.
Предполагается, что первый белок-«зомби» в популяции может появиться случайно, в результате неправильно сложившейся пространственной формы здорового белка PrPС. Эта ошибка, на первый взгляд незначительная, меняет все.
Подобно зомби, ряды которых множатся с каждым укусом, инфекционная форма PrPSc приводит к перерождению нормальных молекул PrPС в новые прионы — процесс развивается как автокаталитическая реакция, продукты которой сами ускоряют ее. Как и зомби, прионы любят «ходить» бесчисленными ордами: частицы PrPSc складываются одна на другую стопками, образуя весьма устойчивые волокна, так что каждый конец такого образования становится центром притяжения для все новых и новых прионов.
«По одиночке, мономерами, PrPSc вообще не встречается, — говорит Илья Баскаков, — они существуют лишь в форме агрегатов-мультимеров. Показано, что самая маленькая частица PrPSc может состоять примерно из шести мономеров, но, вырастая, они доходят до сотен и тысяч единиц». Достигнув больших размеров, белковое волокно разламывается на множество новых фибрилл, каждая из которых становится зародышем новой армии зомби-прионов. Подавленная скоплениями этих бляшек клетка гибнет, а PrPSc распространяются дальше. Уничтожить их крайне непросто: эксперименты показали, что прионы невероятно устойчивы не только к радиации, но и к нагреванию, и даже к действию мощных клеточных ферментов-протеаз.
Достигнув больших размеров, белковое волокно разламывается на множество новых фибрилл, каждая из которых становится зародышем новой армии зомби-прионов. Подавленная скоплениями этих бляшек клетка гибнет, а PrPSc распространяются дальше. Уничтожить их крайне непросто: эксперименты показали, что прионы невероятно устойчивы не только к радиации, но и к нагреванию, и даже к действию мощных клеточных ферментов-протеаз.
Обломки PrPSc могут попадать в организм и извне, через биологические жидкости и ткани больных животных, во время некоторых медицинских процедур и просто с пищей. Большинство переродившихся белковых частиц, видимо, разрушается в желудочно-кишечном тракте, но некоторым удается добраться к месту действия, преодолев даже гемато-энцефалический барьер, стоящий на границе между кровью и тканями мозга. Способность PrPSc проходить сквозь эту весьма надежную преграду остается одной из главных его загадок.
Добрые монстры
Такие свойства характерны не только для PrPС — даже у дрожжей найден свой «прионный белок» Ure2, способный переходить в нестандартную, но крайне устойчивую амилоидную форму. Возможно, это неспроста: «Прионы могут и не быть патогенными, — написал по этому поводу исследователь Рэндал Халфманн, — они могут играть роль „ненуклеиновой“, белковой наследственности у здоровых клеток и организмов». В подтверждение этой идеи Халфманн и его коллеги показали, что дрожжевой белок Ure2, приобретая нестандартную конформацию, влияет на целый ряд сигнальных путей клетки, в том числе и на активность гена FLO11. В свою очередь, синтезируемый на этом гене белок Flo11p необходим клеткам дрожжей для того, чтобы «сцепляться» друг с другом, формируя устойчивые к неблагоприятным условиям пленки. Так «прионное перерождение» белка Ure2 может способствовать адаптации и выживанию клеток.
Возможно, это неспроста: «Прионы могут и не быть патогенными, — написал по этому поводу исследователь Рэндал Халфманн, — они могут играть роль „ненуклеиновой“, белковой наследственности у здоровых клеток и организмов». В подтверждение этой идеи Халфманн и его коллеги показали, что дрожжевой белок Ure2, приобретая нестандартную конформацию, влияет на целый ряд сигнальных путей клетки, в том числе и на активность гена FLO11. В свою очередь, синтезируемый на этом гене белок Flo11p необходим клеткам дрожжей для того, чтобы «сцепляться» друг с другом, формируя устойчивые к неблагоприятным условиям пленки. Так «прионное перерождение» белка Ure2 может способствовать адаптации и выживанию клеток.
Еще одну ложку меда к бочке прионного дегтя добавили работы Джеймса Чена и его коллег. Исследователи показали, что белок MAVS, один из необходимых для работы нашей иммунной системы, переходит в амилоидную прионную форму не во зло, а исключительно во благо. Обнаружив инфицированную определенными вирусами клетку, он «оседает» в ней, и его накопление служит сигналом к усиленному синтезу интерферонов и привлечению макрофагов для уничтожения зараженной клетки вместе со всем ее опасным содержимым.
«На этот счет существует интересная гипотеза, которая указывает на то, что такие амилоидные формы могут иметься вообще у всех белков, что это общее свойство полипептидных цепей, — продолжает Илья Баскаков. — Мало того, такие структуры наиболее стабильны. Поэтому высказано предположение о том, что именно в таких формах белки могли существовать в „пребиотическом супе“, в котором некогда проходила химическая эволюция молекул и зарождалась жизнь».
Действительно, некоторые эволюционисты полагают, что амилоидные конформации белков обладают всеми ключевыми способностями, необходимыми для роли «предков жизни». Они способны изменяться и размножаться, выполнять определенные функции, передавая свои особенности следующим поколениям. Если эта идея справедлива, то мы живем в очень странном мире, где все живое появилось на свет от странных и опасных белков, которые сегодня мы можем воспринимать не иначе как безжалостных зомби.
Симптомы, причины, лечение и профилактика
Прионные заболевания представляют собой группу редких нейродегенеративных заболеваний, которые могут поражать как людей, так и животных.
Они вызваны аномальной укладкой белков в головном мозге, в частности, неправильной укладкой прионных белков (PrP).
Это приводит к прогрессирующему ухудшению функций мозга, включая изменения в памяти, поведении и движении. В конце концов, прионные болезни смертельны.
Ежегодно в США регистрируется около 300 новых случаев прионной болезни.
Это может быть:
- , полученные через загрязненное пищевое или медицинское оборудование
- Унаследован с помощью мутаций в ген, который кодирует для PRP
- , где не совместимый PRP -карт. У людей с прионной болезнью неправильно свернутый PrP может связываться со здоровым PrP, что приводит к неправильному свертыванию здорового белка.
Неправильно свернутый PrP начинает накапливаться и образовывать скопления в мозге, повреждая и убивая нервные клетки.
Это повреждение приводит к образованию крошечных отверстий в мозговой ткани, что делает ее похожей на губку под микроскопом.
 (Вот почему вы можете встретить прионные заболевания, называемые «губкообразными энцефалопатиями».)
(Вот почему вы можете встретить прионные заболевания, называемые «губкообразными энцефалопатиями».)Исследователи все еще работают над тем, чтобы больше узнать о прионных болезнях и найти эффективное лечение. Но кое-что они знают.
Читайте дальше, чтобы узнать о различных типах прионной болезни, о способах ее предотвращения и многом другом.
Прионная болезнь может возникать как у людей, так и у животных. Ниже приведены некоторые различные типы прионных болезней. Более подробная информация о каждом заболевании приведена в таблице.
Human prion diseases Animal prion diseases Creutzfeldt-Jakob disease (CJD) Bovine spongiform encephalopathy (BSE) Variant Creutzfeldt-Jakob disease (vCJD) Хроническая истощающая болезнь (ХИБ) Семейная бессонница со смертельным исходом (FFI) Скрейпи Синдром Герстмана-Штраусслера-Шейнкера (СГШ) Feline spongiform encephalopathy (FSE) Kuru Transmissible mink encephalopathy (TME) Ungulate spongiform encephalopathy Human prion diseases
- Creutzfeldt-Jakob disease (CJD).
 Впервые описанный в 1920 году, CJD может быть приобретенным, унаследованным или спорадическим. Большинство случаев CJD являются спорадическими.
Впервые описанный в 1920 году, CJD может быть приобретенным, унаследованным или спорадическим. Большинство случаев CJD являются спорадическими. - Вариант болезни Крейтцфельдта-Якоба (vCJD). Эта форма CJD может быть получена при употреблении зараженного мяса коровы.
- Смертельная семейная бессонница (FFI). FFI влияет на таламус, часть вашего мозга, которая управляет циклами сна и бодрствования. Одним из основных симптомов этого состояния является усиление бессонницы. Мутация наследуется по доминантному типу, а это означает, что больной имеет 50-процентный шанс передать ее своим детям.
- Синдром Герстмана-Штраусслера-Шейнкера (СГШ). ГСС тоже передается по наследству. Как и FFI, он передается доминирующим образом. Это влияет на мозжечок, который является частью мозга, которая управляет балансом, координацией и равновесием.
- Куру. Куру был идентифицирован в группе людей из Новой Гвинеи.
 Болезнь передавалась через форму ритуального каннибализма, при котором поедали останки умерших родственников.
Болезнь передавалась через форму ритуального каннибализма, при котором поедали останки умерших родственников.
Факторы риска этих заболеваний включают:
- Генетика. Если у кого-то в вашей семье есть наследственная прионовая болезнь, вы также подвергаетесь повышенному риску мутации.
- Возраст. Спорадические прионные заболевания, как правило, развиваются у пожилых людей.
- Продукты животного происхождения. Потребление продуктов животного происхождения, зараженных прионом, может привести к передаче вам прионной болезни.
- Медицинские процедуры. Прионные болезни могут передаваться через зараженное медицинское оборудование и нервную ткань. Случаи, когда это произошло, включают передачу через зараженные трансплантаты роговицы или трансплантаты твердой мозговой оболочки.
Прионные болезни животных
- Губчатая энцефалопатия крупного рогатого скота (ГЭКРС).
 Этот тип прионной болезни, обычно называемый «коровьим бешенством», поражает коров. Люди, употребляющие в пищу мясо коров с ГЭКРС, могут подвергаться риску вБКЯ.
Этот тип прионной болезни, обычно называемый «коровьим бешенством», поражает коров. Люди, употребляющие в пищу мясо коров с ГЭКРС, могут подвергаться риску вБКЯ. - Хроническая истощающая болезнь (ХИБ). CWD поражает таких животных, как олени, лоси и лоси. Он получил свое название из-за резкой потери веса, наблюдаемой у больных животных.
- Скрепи. Скрепи — старейшая форма прионной болезни, описанная еще в 1700-х годах. Это влияет на животных, таких как овцы и козы.
- Губчатая энцефалопатия кошек (ГСЭ). FSE поражает домашних кошек и диких кошек в неволе. Многие случаи FSE произошли в Соединенном Королевстве, а некоторые также наблюдались в других частях Европы и Австралии.
- Трансмиссивная энцефалопатия норок (ТМЭ). Эта очень редкая форма прионной болезни поражает норок. Норка — это небольшое млекопитающее, которое часто разводят для производства меха.
- Губчатая энцефалопатия копытных.
 Это прионовое заболевание также встречается очень редко и поражает экзотических животных, родственных коровам.
Это прионовое заболевание также встречается очень редко и поражает экзотических животных, родственных коровам.
Заболевания, вызываемые прионами
Вышеуказанные прионные заболевания — не единственные заболевания, связанные с прионами.
Другие нейродегенеративные заболевания, такие как болезни Альцгеймера и Паркинсона, также связаны с неправильным свертыванием белков в центральной нервной системе. И исследования показали, что некоторые из этих белков с неправильной укладкой могут быть прионами.
Но некоторые ученые считают, что эти белки действуют только как прионы. Они утверждают, что они не могут быть прионами, поскольку болезни, которые они вызывают, такие как болезнь Альцгеймера, не считаются заразными.
Прионные болезни имеют очень длительный инкубационный период, часто порядка многих лет. Когда симптомы развиваются, они прогрессивно ухудшаются, иногда быстро.
Общие симптомы прионной болезни включают:
- трудности с мышлением, памятью и суждениями
- изменения личности, такие как апатия, возбуждение и депрессия
- спутанность сознания или дезориентация
- непроизвольные мышечные спазмы (миоклонус) атаксия)
- проблемы со сном (бессонница)
- затрудненная или невнятная речь
- нарушение зрения или слепота
В настоящее время нет лекарства от прионной болезни.
 Но лечение направлено на обеспечение поддерживающей терапии.
Но лечение направлено на обеспечение поддерживающей терапии.Примеры этого вида ухода включают:
- Лекарства. Некоторые лекарства могут быть назначены для облегчения симптомов. Примеры включают:
– уменьшение психологических симптомов с помощью антидепрессантов или седативных средств
– обезболивание с помощью опиоидных препаратов
– ослабление мышечных спазмов с помощью таких препаратов, как вальпроат натрия и клоназепам - Помощь. По мере прогрессирования заболевания многим людям требуется помощь в уходе за собой и выполнении повседневных дел.
- Обеспечивает увлажнение и питание. На поздних стадиях заболевания может потребоваться внутривенное введение жидкости или зонд для кормления.
Ученые продолжают работать над поиском эффективного лечения прионных болезней.
Некоторые из потенциальных методов лечения, которые исследуются, включают использование анти-прионных антител и «анти-прионов», которые ингибируют репликацию аномального PrP.

Поскольку прионные заболевания могут проявлять симптомы, сходные с другими нейродегенеративными расстройствами, их может быть трудно диагностировать.
Единственным способом подтверждения диагноза прионной болезни является биопсия головного мозга, проводимая после смерти.
Медицинский работник может использовать ваши симптомы, историю болезни и несколько тестов для диагностики прионной болезни.
Тесты, которые они могут использовать, включают:
- МРТ. МРТ может создать детальное изображение вашего мозга. Это может помочь медицинскому работнику визуализировать изменения в структуре мозга, связанные с прионной болезнью.
- Исследование спинномозговой жидкости (ЦСЖ). ЦСЖ можно собрать и протестировать на маркеры, связанные с нейродегенерацией. В 2015 году был разработан тест для специфического выявления маркеров прионной болезни человека.
- Электроэнцефалография (ЭЭГ). ЭЭГ регистрирует электрическую активность в вашем мозгу.
 Аномальные паттерны могут возникать при прионной болезни, особенно при БКЯ, когда можно наблюдать короткие периоды повышенной активности.
Аномальные паттерны могут возникать при прионной болезни, особенно при БКЯ, когда можно наблюдать короткие периоды повышенной активности.
Для предотвращения передачи приобретенных прионных заболеваний был принят ряд мер. Из-за этих упреждающих шагов заражение прионной болезнью через продукты питания или медицинские учреждения в настоящее время крайне редко.
Некоторые из предпринятых превентивных мер включают:
- установление жестких правил ввоза крупного рогатого скота из стран, где встречается коровья энцефалопатия
- запрет на использование частей коровы, таких как головной и спинной мозг, в пищу для людей или животных
- предотвращение с историей или риском заражения прионной болезнью в результате донорства крови или других тканей
- использование надежных мер стерилизации медицинских инструментов, которые вступили в контакт с нервной тканью человека с подозрением на прионную болезнь
- уничтожение одноразовых медицинских инструментов
В настоящее время нет способа предотвратить наследственные или спорадические формы прионной болезни.

Если у кого-то в вашей семье есть наследственная прионная болезнь, вы можете проконсультироваться с консультантом по генетике, чтобы обсудить риск развития этой болезни.
Прионные заболевания представляют собой редкую группу нейродегенеративных заболеваний, вызванных аномально свернутыми белками в головном мозге.
Неправильно свернутый белок образует скопления, которые повреждают нервные клетки, что приводит к прогрессирующему снижению функции мозга.
Некоторые прионные болезни передаются генетически, в то время как другие могут передаваться через зараженные продукты питания или медицинское оборудование. Другие прионные болезни развиваются без какой-либо известной причины.
В настоящее время нет лекарства от прионных болезней. Вместо этого лечение направлено на оказание поддерживающей терапии и облегчение симптомов.
Исследователи продолжают работать, чтобы узнать больше об этих болезнях и разработать потенциальные методы лечения.
Определение, симптомы, лечение и др.

Прионные болезни встречаются редко и возникают из-за белков в мозге, которые «неправильно сворачиваются». Другое название прионной болезни — губчатая энцефалопатия (ТГЭ). Существует много типов прионных болезней, которые поражают как животных, так и людей.
Сворачивание белков — важный процесс в организме. Для правильного функционирования белки должны складываться в определенные трехмерные формы. Когда белок неправильно складывается, он теряет свою структуру и не может функционировать. Неправильно свернутые белки могут вызывать заболевания у людей.
Мозг содержит большое количество нормально свернутых прионных белков, но ученым еще предстоит полностью понять их функцию и почему они неправильно свернуты.
Прионовая болезнь неизлечима. Однако ученые ищут новые подходы к лечению этого состояния.
Читайте дальше, чтобы узнать больше о прионной болезни, включая симптомы, диагностику и лечение.
Прионные болезни — это редкие неизлечимые заболевания головного мозга, поражающие млекопитающих, включая человека.
Иногда терминология для описания прионных болезней может быть непоследовательной. «Прионы» — это болезнетворные агенты, которые могут стимулировать аномальное сворачивание «прионных белков». В своем обычном здоровом состоянии прионные белки обычно присутствуют в головном мозге.
Когда прионные белки начинают аномально складываться и слипаться, образуя амилоидные бляшки, это приводит к повреждению головного мозга.
Болезнетворные прионы могут передаваться различными путями, например, через корм для животных или нестерилизованное медицинское оборудование. Однако важно отметить, что случаи, связанные с оборудованием, имели место до того, как медицинские учреждения внедрили обычные методы стерилизации. По данным Центров по контролю и профилактике заболеваний (CDC), с 19 года не было зарегистрировано ни одного случая использования медицинского оборудования.76.
Некоторые прионные заболевания имеют редкую генетическую связь, например фатальная семейная бессонница.

Когда люди начинают испытывать последствия прионной болезни, их состояние обычно быстро ухудшается. Прионные болезни всегда смертельны.
Несколько типов прионных болезней могут поражать как людей, так и животных. Хотя эксперты определили множество типов, точно неизвестно, подходит ли кто-то к одному точному типу. Вероятно, это связано с отсутствием определенных тестов, которые могли бы подтвердить диагноз или исключить другие.
Прионные заболевания человека
Примеры наиболее распространенных прионных заболеваний включают:
- Болезнь Крейтцфельдта-Якоба (БКЯ): Этот тип БКЯ подразделяется на три типа: семейный, спорадический и приобретенный. Люди наследуют семейный тип, но спорадический тип развивается без каких-либо известных причин. Спорадический CJD является наиболее распространенным типом CJD и, как правило, поражает людей в возрасте около 60 лет. У человека может развиться приобретенная БКЯ после того, как нестерилизованное медицинское оборудование поместило в организм прионы, хотя это случается редко.

- Вариант болезни Крейтцфельдта-Якоба (vCJD): Этот тип CJD представляет собой инфекционное прионовое заболевание, связанное с коровьим бешенством. Люди заражаются им, употребляя в пищу мясо, содержащее белки, из головного или спинного мозга больной коровы. В отличие от спорадической БКЯ, вБКЯ чаще поражает молодых людей.
- Семейная бессонница со смертельным исходом: Этот тип — обычно наследственный — связан с наследованием атипичной формы гена, кодирующего прионные белки. Редко это заболевание возникает спорадически. В течение болезни люди спят все меньше и меньше. Это может привести к ухудшению психического состояния и физическим симптомам.
- Синдром Герстмана-Штраусслера-Шейнкера: Это генетическое заболевание, поражающее прионные белки в мозжечке. Мозжечок — это часть мозга, которая, помимо прочих функций, контролирует движение и равновесие.
Прионные болезни животных
Примеры распространенных прионных болезней животных включают:
- губчатая энцефалопатия крупного рогатого скота, также известная как коровье бешенство, поражающая крупный рогатый скот
- хроническая истощающая болезнь, поражающая оленей и лосей
- скрепи, поражающая овец
В настоящее время ученые не до конца понимают, что вызывает неправильную укладку прионных белков.
 Кроме того, прионные белки могут неправильно сворачиваться в течение многих лет, прежде чем у человека появятся симптомы.
Кроме того, прионные белки могут неправильно сворачиваться в течение многих лет, прежде чем у человека появятся симптомы.Факторы риска
Прионные болезни встречаются редко. CDC сообщает об 1 случае CJD на миллион человек ежегодно.
Однако факторы риска прионной болезни могут включать:
- Семейный анамнез прионной болезни, особенно фатальной семейной бессонницы.
- Употребление в пищу или контакт с мясом, содержащим белки головного или спинного мозга больной коровы, особенно с вБКЯ.
- Передача — например, приобретение болезни при медикаментозном или хирургическом лечении.
- Возраст, особенно для CJD.
Симптомы прионной болезни могут различаться в зависимости от типа неправильно свернутого прионного белка. Различные прионные белки могут быть нацелены на определенные области мозга. Следовательно, симптомы могут отражать повреждение областей мозга прионами.
Например, в случаях семейной бессонницы со смертельным исходом человек не может спать и обычно видит яркие сны в дополнение к изменениям температуры тела.
 По мере прогрессирования болезни они спят все меньше и меньше.
По мере прогрессирования болезни они спят все меньше и меньше.В качестве альтернативы, у человека с CJD могут сначала развиться симптомы, подобные слабоумию, и возникнуть проблемы с равновесием. Они также могут сообщить:
- головокружение
- головные боли
- утомляемость
Поскольку прионные заболевания поражают головной мозг, у людей обычно наблюдается:
- changes in gait and walking
- hallucinations
- muscle stiffness
- confusion
- fatigue
- speech difficulties
- atypical jerking movements
- rapid-onset dementia
Usually, it takes a long time — even years — симптомы проявляются после того, как прионные белки начинают неправильно свертываться.
Сопутствующие состояния
Некоторые симптомы прионной болезни могут совпадать с другими заболеваниями, например:
- Болезнь Альцгеймера
- Болезнь Паркинсона
- Деменция с тельцами Леви
- хроническая травматическая энцефалопатия, возникающая после повторной травмы головного мозга
исключить их.
 Это потому, что они более распространены, чем прионные болезни.
Это потому, что они более распространены, чем прионные болезни.Прионные болезни невероятно сложны. Каждое заболевание имеет свои собственные диагностические критерии, обычно состоящие из различных медицинских тестов и физических обследований.
Сюда могут входить:
- Анализы крови: Обнаруживает наличие прионов в крови. Люди также могут получить прототип тестов, поскольку прионные заболевания очень редки.
- Генетические тесты: Определяет, есть ли у кого-то варианты генов.
- МРТ: Обнаруживает изменения в структуре мозга.
- Люмбальная пункция: Скрининг спинномозговой жидкости на определенные маркеры CJD.
- Электроэнцефалограмма: Измеряет любые изменения мозговых волн.
Некоторым людям может потребоваться биопсия головного мозга, когда врач использует небольшую иглу для взятия образца ткани.
 Обычно это делается под общим наркозом.
Обычно это делается под общим наркозом.Биопсия головного мозга является процедурой высокого риска, поскольку хирурги удаляют часть ткани головного мозга. Кроме того, в случае прионной болезни существует риск для любого, кто работает с взятой пробой. Лучший способ подтвердить диагноз при таких состояниях, как CJD, — это аутопсия, а не биопсия.
Врачи также могут запросить чувствительный диагностический тест для обнаружения некоторых прионов или прионных белков. Например, в случаях CJD они могут использовать анализ, вызывающий конверсию в режиме реального времени, или анализ RT-QuIC. Этот метод может усиливать неопределяемые уровни прионов. До этого анализа риск ложноотрицательного результата был выше.
Люди должны иметь в виду, что анализ RT-QuIC не является широко доступным.
В настоящее время не существует методов лечения прионной болезни. Подходы к лечению могут быть сосредоточены на лечении симптомов.
Однако ученые исследуют влияние различных молекул, которые могут ингибировать образование прионов.

- Creutzfeldt-Jakob disease (CJD).